
{kind=link}
{kind=link}
羊草叶绿体非编码区多态性标记筛选及群体遗传多样性
[杨艳婷1  , 侯向阳
, 侯向阳1 , 魏臻武2 , 乔志宏1 , 常春1 , 任卫波1 , 武自念1, * ]
, 侯向阳]
|
|


作者简介:杨艳婷(1990-),女,河南商丘人,在读硕士。E-mail: tingyy@outlook.com
为筛选多态性丰富的羊草叶绿体非编码区片段,本研究以9个不同地理位置的羊草居群为材料,通过对12个叶绿体非编码区DNA片段测序及其序列间变异分析试图从中找出有遗传差异的叶绿体DNA(cpDNA)片段,并利用有多态性的cpDNA片段进行遗传多样性分析。结果表明,序列 ndhF-rpl32、 trnL-trnF、 trnC-ycf6、 aptI-aptH具有比较丰富的变异,可作为下一步研究野生羊草群体遗传学和谱系地理学较为理想的分子标记。在37个羊草个体4条非编码区片段的合并序列中,共检测到15种单倍型,单倍型多态性( Hd)为0.928,核苷酸多态性( Pi)为0.00101;遗传分化指数( Fst)为0.58884,基因流( Nm)为0.17,基因流较小;中性检验Tajima’s D(-1.08542)和Fu’s Fs(-5.301)均为负值,且差异不显著( P>0.10),推测羊草遵循中性进化理论,可能经历过种群扩张;AMOVA结果显示,65%的分子变异出现在居群间,35%出现在居群内;Mantel检验得到,遗传距离与地理距离具有显著相关性( r=0.449, P<0.05);居群分化值 Nst 0.386(0.1865)大于 Gst 0.234(0.1506),差异显著( P<0.05),表明羊草居群存在分子谱系地理结构。通过系统发育树分析得到,羊草15个单倍型分为两大分支,H2和H10聚为一支,它们与别的居群个体亲缘关系较远,其余单倍型聚为另一支;单倍型网络图显示,H2和H12、H10和H12亲缘关系较远,与系统发育树分析结果基本一致。本研究为羊草的种质资源保护、谱系地理学研究等工作奠定了基础。
, HOU Xiang-yang
The aim of this research was to screen for and classify polymorphic loci of chloroplast non-coding fragments in Leymus chinensis. The study evaluated 9 L. chinensis populations from different geographical locations to identify polymorphic loci from 12 chloroplast DNA (cpDNA) non-coding fragments, and then genetic diversity of the polymorphic cpDNA fragments was analyzed. The results showed that the polymorphic loci of ndhF-rpl32, trnL-trnF, trnC-ycf6 and aptI-aptH were rich, and they had a relatively fast evolution rate. Hence, they could be regarded as ideal molecular markers for future research on population genetics and phylogeography. Based on combined sequences there were 15 haplotypes identified. The haplotype diversity ( Hd) and nucleotide diversity ( Pi) were 0.928 and 0.00101 respectively. The genetic differentiation coefficient Fst was 0.58884 with the low gene flow ( Nm=0.17). Tajima’s D and Fu’s Fs values of neutrality tests were negative (Tajima’s D=-1.0854; Fu’s Fs=-5.301), indicating that L. chinensis had experienced historical population expansion. Analysis of molecular variance showed that most of genetic variation was distributed among populations. The Mantel test showed a significantly positive correlation between genetic and geographical distance ( r=0.449, P<0.05). The genetic differentiation coefficients Nst (0.386)> Gst (0.234) implied a significant phylogeographic structure for the populations. The neighbor-joining tree based on combined sequences showed that all haplotypes divided into two major clades, H2 and H10 clustered into a category. The network for cpDNA haplotypes based on combined sequence data showed that the relationships among H2 and H12, and H10 and H12 were further than others, which was similar to the pattern identified by the neighbor-joining tree. These results will be useful for future research on germplasm resources and phylogeography.
羊草(Leymus chinensis)是欧亚大陆草原具有优势的牧草和生态草之一, 高产抗逆性强, 分布范围广, 为大多数畜牧动物所喜食, 是恢复退化天然草原和建设优质人工草地极其重要的草资源[1]。羊草是欧亚大陆草原区东部草甸草原及典型草原的重要建群种, 作为我国重要的牧草和生态草, 羊草能适应多种环境条件, 具有极丰富的遗传多样性, 羊草种群之间也产生了不同的遗传特征[2, 3]。探讨不同地理种群羊草的遗传特性对于研究羊草分化起源及育种等具有重要的理论价值和实践意义。
叶绿体基因是研究植物谱系地理学、系统学及种群历史的首选材料。植物体内叶绿体基因进化速率较慢, 仅为核基因的一半, 且叶绿体DNA(chloroplast DNA, cpDNA)为母系遗传, 不会受基因重组的干扰, 进化路线相对独立[4, 5, 6]。叶绿体DNA非编码区的核苷酸替换速率相对较高, 有研究指出, 叶绿体非编码区因受到的限制较少, 能产生比编码区更多的变异位点, 可以提供较多的具有系统学意义的信息位点[7, 8, 9]。目前, atpB-rbcL、trnL-trnF等叶绿体基因非编码区广泛应用于物种遗传多样性及谱系地理学研究。近年来, 草本植物基于叶绿体非编码区的研究也越来越多, 吴娟子等[10]利用叶绿体基因非编码区trnT-trnF序列分析了中国互花米草(Spartina alterniflora)不同种群的遗传结构, 赵艳宁[11]利用cpDNA非编码区trnL-trnF和trnH-psbA对短花针茅(Stipa breviflora)11个居群165个个体材料进行了分子谱系地理学研究, 杨青松等[12]用cpDNA psbA-trnH序列对不同海拔的血满草(Sambucus adnata)进行了遗传结构分析。在整个叶绿体基因组中, 非编码区在核苷酸替代或插入/缺失突变上, 都表现出比编码区更快的进化速率, 受越来越多的研究者青睐。
目前, 人们对羊草叶绿体基因的研究主要集中在羊草与近缘种之间的系统发育学研究上。Sun[13]利用rbcL基因分析了披碱草属与含羊草在内的小麦族18个物种24个个体之间的系统进化关系, 构建系统发育树, 结果显示, 羊草与新麦草(Psathyrostachys fragilis)之间的亲缘关系较近。羊草是赖草属的一种, 利用叶绿体基因间隔区进行属内系统发育学的研究也有报道。Liu等[14]利用核基因ITS序列和叶绿体基因间隔区trnL-trnF序列对赖草属及近缘种13个物种57个个体进行了系统进化分析, 也得到羊草与新麦草有较近的亲缘关系。Guo等[15]利用叶绿体基因间隔区trnQ-rps16序列分析了赖草属25个物种36个个体之间的系统发育关系, 并得到trnQ-rps16序列很有可能是区分赖草属进化关系的最佳选择。利用cpDNA非编码区序列研究羊草种内遗传多样性及进化方面的报道较少, 因而筛选出合适的多态性基因对进一步探讨羊草种内系统进化、谱系地理关系极为重要。
本研究以9个不同地理位置的羊草居群为材料, 分别对12个叶绿体非编码区DNA片段进行测序, 通过序列间变异程度分析, 试图从中找出变异相对丰富的cpDNA片段, 并对所选材料的遗传多样性、遗传分化、基因流、系统发育树以及单倍型网络图进行分析, 为研究羊草群体遗传学, 探讨系统发生关系和进行谱系地理学研究, 估测遗传多样性中心进而保护羊草种质资源, 培育高产、抗旱、耐盐碱优良羊草品种等方面的工作提供依据。
2016年8月在中国农业科学院草原研究所沙尔沁试验基地羊草种质资源圃采集9个羊草居群37个个体鲜嫩叶片后液氮速冻(表1), 于-80 ℃冰箱保存备用。9个羊草居群的材料是来自不同地理位置的野生种质, 每个居群的个体位置均以该GPS点为中心采样, 个体之间相距40 m, 取整株植株后带回, 种植在羊草种质资源圃。
| 表1 羊草9个不同地理位置的居群信息 Table 1 The information of 9 different geographic populations in L. chinensis |
采用植物基因组DNA提取试剂盒提取羊草全基因组DNA, 溶于Tris+EDTA(TE)缓冲液后在0.8%琼脂糖凝胶中电泳检测并拍照, 存于-20 ℃冰箱备用。植物基因组DNA提取试剂盒(DP-305)购自天根生化科技北京有限公司。
PCR扩增反应总体积为50 μ L:2* Taq PCR MasterMix 25 μ L, 引物F、R各3 μ L(10 μ mol· L-1), ddH2O 9 μ L, DNA 10 μ L(约200 ng)。扩增程序为:94 ℃预变性5 min; 94 ℃变性45 s, 43~60 ℃退火45 s, 72 ℃延伸1 min, 共35个循环; 72 ℃再延伸10 min; 10 ℃保存。PCR扩增产物在0.8%琼脂糖凝胶中电泳检测并拍照, PCR扩增产物送往上海生工生物工程股份有限公司进行双向测序, 测序引物与PCR反应引物相同。引物由上海生工生物工程股份有限公司合成(表2)。2* Taq PCR MasterMix购自天根生化科技北京有限公司。
| 表2 12条叶绿体非编码区片段引物信息 Table 2 The primer of 12 different chloroplast non-coding fragments |
使用Seqman软件读取序列, 去除两端峰图信号杂乱的低质量碱基, 并进行手工拼接和人工校对。用MEGA 5.0软件进行序列比对, 比对结果利用Dnasp 5.10软件统计可变位点和简约性信息位点; 采用MEGA 5.0软件构建系统发育树, 并利用Network 5.0软件进行单倍型网络图分析; 利用GenAlEx 6.5进行分子变异分析(analysis of molecular variance, AMOVA)和Mantel检验; 用PERMUT软件来计算居群分化指数Gst与Nst值, 并用1000次置换进行检验; 利用Dnasp 5.10进行中性检验, 获得Tajima’ s D和Fu’ s Fs值。
9个羊草居群37个个体的12条叶绿体非编码区序列全长8499 bp, 插缺和变异位点数为99个, 占序列长度的1.16%; 变异位点数为28个, 简约性信息位点16个, 插缺位点12处含2处长碱基插缺(表3)。序列ndhF-rpl32变异位点数最多, 简约性信息位点5个, 分别发生在105、243、249、363和368 bp处, 其次是序列trnL-trnF、trnC-ycf6、aptI-aptH, 均有2个简约性信息位点, 各序列变异情况不同, 整体来看所选羊草叶绿体非编码区序列变异率不高, 仅为0.33%。
| 表3 12个叶绿体非编码区片段多态位点统计 Table 3 The polymorphism of 12 cpDNA non-coding fragments |
所选羊草材料不同叶绿体非编码区片段的遗传多样性不同, 4条非编码区片段合并后全长3075 bp, 单倍型多态性(Hd)为0.928, 核苷酸多态性(Pi)为0.00101。片段ndhF-rpl32具有相对较高的遗传多样性水平, 所有居群的单倍型多态性(Hd)为0.778, 核苷酸多态性(Pi)为0.00227, 核苷酸平均差异数(K)为1.595(表4), 在4条基因片段中均最高。单倍型多态性(Hd)和核苷酸多态性(Pi)最小的序列是trnL-trnF, 分别为0.00023、0.158。4条非编码区片段合并后共检测到15种单倍型, 单倍型多样性方差和标准差分别是0.00035、0.019。
| 表4 基于4条叶绿体非编码区片段羊草的遗传多样性和单倍型多样性分析 Table 4 The genetic diversity and haplotype diversity analysis in L. chinensis based on 4 cpDNA non-coding fragments |
4个叶绿体非编码区片段的Tajima’ s D值均为负值(表4), 且均不显著(P> 0.10); 序列trnL-trnF的D值最低, Tajima’ s D=-1.08228, 序列trnC-ycf6和aptI-aptH的Tajima’ s D值均为-0.69971。序列合并后, Tajima’ s D值为-1.08542, Fu’ s Fs值为-5.301, 且不显著(P> 0.10), 说明羊草在进化的过程中遵循中性进化模式, 推测可能经历过群体扩张。
4条叶绿体非编码区序列合并后羊草9个居群之间的多态性表现不同(表5)。居群Pop3、Pop7的遗传多样性水平最丰富, 居群Pop3的核苷酸多态性(Pi=0.00153)和平均核苷酸差异(K=4.6667)最高, 居群Pop7具有的单倍型数目最多, 且核苷酸多态性(Pi=0.00105)和平均核苷酸差异(K=3.2)也相对较高。居群Pop1、Pop5、Pop8、Pop9具有单一的一种单倍型, 核苷酸多态性和单倍型多态性均为0。
| 表5 基于合并序列9个羊草居群的遗传多样性和单倍型多样性分析 Table 5 The genetic diversity and haplotype diversity analysis of different populations in L. chinensis based on combined sequence |
由单倍型分布可以看出, 15种单倍型中只有H5被居群Pop3和Pop7共享, H7被居群Pop2、Pop4、Pop7共享, 其他的单倍型都是地域特异性地分布在一个地点的群体中, 为各居群所特有, 说明各居群较为独立, 可能存在潜在的质粒水平的隔离[27]。
通过AMOVA 分析可知, 羊草居群65%的遗传变异存在于群体间, 35%存在于群体内(表6, P< 0.01), 居群间遗传变异大于居群内; 遗传分化系数Fst=0.58884, 说明羊草居群间存在较大的遗传分化, 居群间变异是羊草的主要变异来源。根据基因流(Nm)与Fst之间的关系:Nm=(1-Fst)/4Fst[28], 得到Nm为0.17, 基因流较小。
| 表6 基于合并序列羊草居群内与居群间的分子变异分析 Table 6 AMOVA for different populations in L. chinensis based on combined sequence |
羊草各居群间的遗传距离较小, 居群Pop3 和Pop8、Pop8和Pop9遗传距离达到最大, 为0.002, 其他各居群之间多为0.001(表7)。居群之间的遗传分化指数表明, 居群Pop3和Pop7没有表现出遗传分化(-0.093), 居群Pop2和Pop6(0.214)、Pop3和Pop6(0.154)、Pop4和 Pop7(0.178)、Pop6和 Pop7(0.170)遗传分化较大, 其他各居群之间均为高度分化[29](表7), 同时较小的基因流(0.17)也加剧了居群之间的遗传分化。
| 表7 基于合并序列不同羊草居群的遗传距离和遗传分化指数 Table 7 Genetic distance and pairwise fixation indices of genetic variation of different populations in L. chinensis based on combined sequence |
PERMUT软件分析结果显示, 羊草所选分布区居群内的平均遗传多样性Hs为0.732(0.1401), 居群间总遗传多样性Ht为0.956(0.0253), 总的居群分化值Nst 0.386(0.1865)大于Gst 0.234(0.1506), 差异显著(P< 0.05), 表明羊草居群存在分子谱系地理结构; 通过Mantel检验发现, 各居群之间的遗传距离与地理距离存在显著相关性(r=0.449, P< 0.05)。
基于邻接法(Neighbor-Joining, NJ)构建羊草不同单倍型之间的系统发育树(图1)。所选37个羊草个体15个单倍型分为两大分支, 其中单倍型H2和H10聚为一大分支, H3和其余单倍型为另一分支; 除H2、H10和H3以外的单倍型又分为3小分支, H5、H8和H13聚为一支, H9单独聚为一支, 其余聚为另一支, 其中H4和H6、H11和H12的居群亲缘关系较近, 各聚为一支。
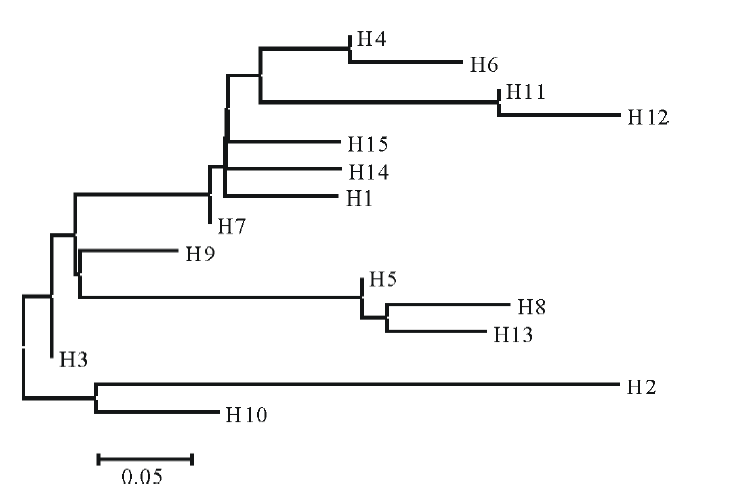 | 图1 基于合并序列不同羊草居群的系统发育NJ树Fig.1 NJ tree of different populations based on combined sequence |
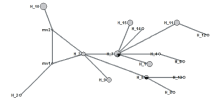
运用中介邻接网络(Median-Joining network, MJ)算法, 构建羊草不同地理位置种质cpDNA单倍型间的关联图(图2)。单倍型和中介矢量载体的名称分别表示为H-和mv。单倍型H3位于分支的躯干上, 通过mv1和mv2将H2和H10、H2和H3连接起来。H2和H3、H3和H10的居群亲缘关系较远; H11和H12、H4和H6的居群亲缘关系较近, 与H1、H14和H15聚在H7节点上; H8、H13与H5的亲缘关系较近; H5、H7和H9共同聚在H3节点上, 亲缘关系较近(图2); 这与系统发育树分析结果基本一致。
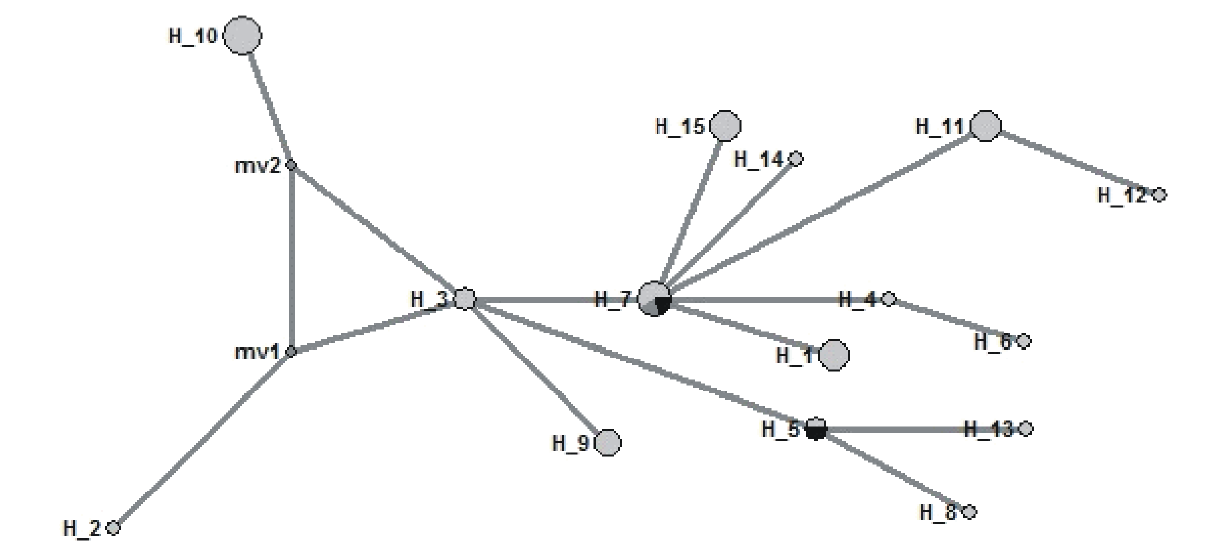 | 图2 基于合并序列的单倍型网络图 圆圈面积大小与单倍型频率呈正比, 同一单倍型圆圈中的不同颜色代表不同的群体, mv1和mv2表示假定的单倍型节点。Fig.2 The network for cpDNA haplotype based on combined sequence The area of the circle is proportional to the frequency of haplotype; the colors in the same circle represent different populations. |
牧草叶绿体基因组大小一般为120~170 kb, 基因组通常包括120~130个基因, 其基因容量和基因顺序非常保守[28]。本研究羊草叶绿体基因组12条非编码区片段共检测到40个变异位点, 其中插入/缺失变异位点12个占总变异的30%。有研究发现叶绿体基因组核苷酸变异中替换约占70%, 插缺突变约占30%, 或替换发生的频率是插缺变异发生频率的3倍[30], 本研究所得的结果与前人研究大致相同。目前对插入/缺失产生的机制和过程还不是十分清楚, 插入/缺失变异的产生过程比核苷酸替代要复杂很多, 由于测序产生的误差和基因组信息的不全, 导致插入/缺失突变很难被准确地检测出来[31], 故插缺变异的统计可能存在误差。
12条叶绿体非编码区片段在所选羊草材料上检测到不同程度的变异, 可能是由各片段进化速率不同导致的[23]。其中序列ndhF-rpl32、trnL-trnF、trnC-ycf6、aptI-aptH变异相对较丰富, 在羊草中的进化速度相对较快, 是低水平分子研究较好的选择[32], 这与前人的研究结果基本一致[17, 19]。基因ndhF-rpl32序列位于小单拷贝SSC区, 有相对较丰富的变异位点, 检测到5处简约性信息位点, 推测其丰富的变异可能是因为在叶绿体基因组DNA复制过程中小单拷贝区的位置较易引入突变。psbA-matK、rpl20-rpl12、trnS-trnG、atpB-rbcL、trnF-ndhJ序列检测到的信息位点较少, 而刘静等[33]利用叶绿体基因间隔区atpB-rbcL序列对小麦族不同属的植物进行序列变异分析, 检测到了较多的简约性信息位点, 约占2.5%。本研究的结果与之不同, 原因可能是不同序列片段在不同类群的物种中得到的变异情况不同, 这几个序列片段进化速率在羊草中相对较慢。
一般来说, 物种遗传多样性受其生存环境、自然进化和本身适应能力等因素影响。通过对9个不同地理分布羊草居群的遗传多样性进行分析, 共鉴别出15种单倍型, 羊草总体上呈现出较低的遗传多态性(Hd=0.928, Pi=0.00101); Nm值(0.17)小于1, 表明羊草各居群的基因交流较小。生殖方式为异交的物种遗传多样性一般小于等于0.1, 羊草居群的核苷酸多态性指数(Pi)仅为0.00101, 远低于0.1, 可能是因为羊草为克隆植物, 主要通过无性系繁殖, 有性生殖能力低下。中性检验Tajima’ s D值和Fu’ s Fs值均为负值, 在P> 0.10水平上不显著, 说明羊草在进化的过程中遵循中性进化模式, 可能经历过种群扩张, 导致其遗传多样性较低[34]。羊草单倍型多态性和核苷酸多态性在每个种群间表现不同, 推测可能是由于羊草分布范围较为广泛, 各种群所处自然环境条件不同, 经过生态选择、自然选择导致不同地理种群羊草的遗传多样性有所不同。刘惠芬等[35]对来自内蒙古草原不同生境8个羊草种群进行RAPD技术分析得到, 较小地理范围内羊草的遗传分化程度较小, 其遗传分化主要是由环境的异质性所引起, 与其生境间的相似度相关。任文伟等[36]也得到羊草的变异和分化是多种生态因子综合作用的结果(如温度、海拔、经纬度、土壤类型等), 钱吉等[37]发现水分是影响羊草种群间遗传变异和生态型分化的最主要的因子, 环境差异是引起羊草遗传变异的主要因素。本研究种群Pop3位于山西运城市, 是地处黄土高原的山地, 种群Pop7位于内蒙古自治区通辽市, 地貌类型以沙丘、沙地为主要特征, Pop3、Pop7种群的遗传多样性水平最丰富, 可能与羊草不同地理种群的生活环境差异较大有关。
植物居群的遗传结构受群体进化历史、基因流等影响, 群体进化历史一般通过遗传分化指数(Fst)来反映, 其大小可在一定程度上揭示种群间基因流和遗传漂变的程度[38]。一般来讲, 当Fst< 0.05, 群体间没有遗传分化; 0.05< Fst< 0.15, 群体分化程度中等; 0.15< Fst< 0.25, 群体间高度分化; Fst> 0.25, 群体间分化程度非常高[29]。羊草居群遗传分化指数(Fst)为0.58884, 说明群体间遗传变异较大, 分化程度较高。羊草为兼性生殖植物, 有一定异交生殖能力, 本研究所选羊草材料地理距离较远, 发生基因交流的可能性较小, 这也加剧居群间遗传分化的增大; 各居群所处环境的不同, 也是产生居群间遗传变异的原因。AMOVA分析显示羊草分子变异主要表现在居群间(65%), 且达到了显著差异(P< 0.01), 这与前人研究结果不同。Gong等[39]和Wang等[40]分别使用AFLP、RAPD分子标记对羊草进行遗传变异分析, 均得到主要变异是发生在种群内。叶绿体DNA比核DNA序列相对保守, 变异速率仅次于核基因组。另外, 使用多个叶绿体分子标记相联合进行分析的方法比使用单一分子标记所能提供的有效信息更为全面可靠[41]。本研究选取4个遗传变异相对丰富的叶绿体分子标记进行遗传变异分析, 提高了遗传多样性检测的灵敏度, 更加客观地反映了羊草各地理居群的遗传结构。
本研究从羊草12条叶绿体非编码区中筛选出ndhF-rpl32、trnL-trnF、trnC-ycf6、aptI-aptH 4条序列变异相对较高, 可作为下一步探讨羊草野生群体谱系地理关系与进化历程理想的分子标记; 羊草遗传多样性水平较低(Hd=0.928, Pi=0.00101), 基因流较小(Nm=0.17), 遗传分化程度较高(Fst=0.58884), 遵循中性进化理论, 可能经历过种群扩张; 各居群遗传距离与地理距离相关性显著, 存在分子谱系地理结构。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|