

{kind=link}
{kind=link}
{kind=link}
{kind=link}
基于转录组测序分析象草木质素合成的研究
[吴娟子1, 2, 3  , 钱晨
, 钱晨1, 2, 3 , 刘智微1, 2, 3 , 潘玉梅1, 2, 3 , 钟小仙1, 2, 3, * ]
, 钱晨]
|
|


作者简介:吴娟子(1977-),女,湖北京山人,副研究员,博士。E-mail: jzwu2014@jaas.ac.cn
采用高通量测序技术Illumlna HiSeq 2000对高木质素的象草品系eg7和低木质素的象草品系eg87(对照)茎组织进行转录组比较测序。测序获得了169630902个序列读取片段(reads),包含13788439920 nt碱基信息。对reads进行序列组装,获得87641个单基因簇(unigene),平均长度580 nt。从长度分布、GC含量等方面对unigene进行评估,数据显示测序质量好,可信度高。将获得的unigene与Nr、Nt、Swiss-Prot、COG、GO和KEGG数据库进行序列同源性比较和功能分析,62557个unigene与其他生物的已知基因具有不同程度的同源性,象草与高粱序列同源性最高。共鉴定出33323个差异表达基因,其中上调基因9704个(29.12%),下调基因23619个(70.88%);GO分析显示39968个unigene归为54个功能类别,大量unigene与细胞进程、代谢过程、催化活性等相关;KEGG pathway分析富集得到127条代谢通路,包括光合作用、betalain生物合成、苯丙烷类代谢、苯丙氨酸代谢等,苯丙烷类代谢途径差异基因富集程度高、差异基因数目最多,达285条,该途径中64条木质素单体合成酶基因表达上调,79条ClassⅢ型植物过氧化物酶基因表达下调、22条上调。挑选9个差异基因进行qRT-PCR验证,9个基因的表达趋势与高通量测序结果一致。为象草的分子生物学研究提供了宝贵的基因组数据,对于了解象草茎生物合成与木质素调控基因挖掘和多用途定向育种具有指导意义。
Elephant grass ( Cenchrus purpureus) is an excellent forage crop and a promising lignocelluloses energy crop; its stem characteristics are very important for forage and feedstock quality. In order to understand the gene expression profile of elephant grass stem in the absence of a reference genome, we performed de novo transcriptome sequencing and compared the transcriptome of stems with a high lignin content plant (HLCP) eg7 and a low lignin content plant (LLCP) eg87 in elephant grass using Illumina HiSeq 2000 platform. A total of 169630902 high qualities reads were obtained, which were then assembled into 87641 unigenes with an average length of 580 nt. All of the 87641 unigenes were compared against the non-redundant protein database (Nr), non-redundant nucleotide database (Nt), swisssprot protein sequence database (Swiss-Prot), gene ontology (GO), cluster of orthologous groups (COG) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) databases for functional annotation; a total of 62557 unigenes were annotated. Most homologous sequences were found between elephant grass with sorghum. 33323 unigenes showed significant differences in expression between eg7 and eg87 (control), 9704 (29.1%) genes were up-regulated and 23619 (70.9%) were down-regulated. GO and KEGG pathway analysis revealed that the genes related to photosynthesis-antenna proteins, photosynthesis, betalain biosynthesis, phenylpropanoid biosynthesis are genes of interest. 9 monolignol biosynthesis-related genes and 8 ClassⅢ peroxidase genes were selected as candidate genes. qRT-PCR analysis showed that the expression of 9 selected unigenes were consistent with the transcriptome data. Our study presented comprehensive transcriptomic data and gene function analysis of C. purpureus stem, providing a valuable resource for future studies of plant breeding in the genus Pennisetum and comparative genome analysis for C4 grasses.
象草(Cenchrus purpureus)是多年生禾本科C4植物, 是世界上生物量最高的草本植物, 年生物量高达45000 kg· h
Illumlna RNA-Seq是第二代转录组测序技术, 具有高通量、低成本的特点, 并且不需要相应的测序参考基因组信息, 对于许多缺乏基因组信息的物种而言, 其具有明显的优势[8]。目前, 转录组测序在紫花苜蓿(Medicago sativa)[9]、白三叶(Trifolium repens)[10]、草地早熟禾(Poa pratensis)[11]、羊草(Leymus chinensis)[12]等牧草研究中都有应用。
本研究在前期系统评价90份象草资源的农艺性状和其中73份资源茎秆细胞壁组成的基础上[13], 挑选了酸性木质素含量最高的eg7和最低的eg87两份材料, eg7和eg87的酸性木质素含量分别为16.9%和5.7%, 对其进行茎转录组测序和分析, 以期为象草的分子生物学研究提供宝贵的基因组数据, 并为进一步挖掘象草木质素合成关键基因奠定基础。
2014年5月4日, 从江苏省农业科学院国家牧草育种创新基地大棚内取越冬保存的象草eg7和eg87根茎, 分株移栽于江苏省农业科学院大田, 供试土壤属粘性马肝土, 肥力中等, 整个试验地肥力均匀。8月6日, 选取生长94 d后、健康的茎杆中部的茎段组织(从基部起第7和第8个茎节之间的茎段组织), 迅速将其放入锡箔纸内, 立即经液氮速冻后保存于-80 ℃备用。每个材料选取3个健康单株, 用于总RNA提取, 等量混合株系内的RNA样品。
10月18日, 收获象草整株茎秆, 剪成10~15 cm茎段, 105 ℃杀青30 min, 70 ℃烘干至恒重。烘干的茎粉碎, 过80目(0.18 mm)筛, 冷却后于密封袋中保存, 待进一步分析。采用范氏洗涤纤维法测定纤维素(cellulose, CEL)、半纤维素(hemicelluloses, HMC)和酸性木质素(acid detergent lignin, ADL)含量[14, 15]。
象草茎段样品送华大基因公司进行转录组测序。利用TRIzol法提取茎段总RNA, RNA纯化后利用NanoDrop和Agilent 2100检测RNA质量, 质量合格后建立测序文库并利用Illumina HiSeq2000平台测序。
获得原始测序数据后, 去除接头序列、低质量序列和N(不确定碱基)比例大于0.01%的序列, 将过滤后的clean data利用SOAP de novo软件进行序列拼接, clean read拼接成重叠群(Contig)后进一步组装成unigene[16]。
利用Blastx和Blastn程序将组装获得的象草unigene与蛋白质、核酸数据库进行比对(e-value< 0.00001), 这些数据库包括非冗余蛋白数据库(non-redundant protein database, Nr)、蛋白质序列数据库(swissprot protein sequence database, Swiss-Prot)、东京基因与基金组百科全书(kyoto encyclopedia of genes and genomes, KEGG)和蛋白质直系同源数据库(cluster of orthologous groups, COG)、非冗余核酸序列数据库(non-redundant nucleotide database, Nt)[17, 18, 19, 20]。选取最佳的比对结果用于基因的功能注释并确定基因的方向。
以eg87为对照、eg7为处理组对所有unigene进行差异表达分析, 找出在两样本间表达量不同的基因。为了确定两个文库之间差异表达的基因, 使用RPKM(每百万reads中来自于某基因每千碱基长度的reads数, reads per kilobases per million mapped reads)方法分析所有组装的unigene的转录丰度, RPKM方法消除了不同基因长度和测序水平对基因表达计算的影响[21]。用错误发现率(false discovery rate, FDR)(FDR≤ 0.001)和两个样本的RPKM比值(|log2 Ratio|≥ 1)作为差异基因的筛选条件。根据比对Nr数据库获得的信息, 使用Blast2GO软件[16]得到差异表达的unigene的基因本体论 (gene ontology, GO)条目, 然后用WEGO软件对这些基因进行GO功能分类统计, 同时利用KEGG数据库比对进行功能分类和代谢途径分析。
分别提取、纯化eg7和eg87两个材料茎段的RNA, 采用RevertAid First Strand cDNA Synthesis Kit (Fermentas)合成cDNA第一链。选取9条木质素单体合成候选unigene, 象草elongation factor 1a (CpEF1a)和actin (CpActin)作为内参基因, 设计qRT-PCR引物, 引物序列见表1。利用LightCyclerR480 fluorescence real time PCR machine (Roche)完成荧光定量PCR实验, PCR程序为:95 ℃预变性10 min; 40个循环(95 ℃变性15 s, 60 ℃退火20 s, 72 ℃延伸40 s, 80 ℃ 1 s收集荧光, 读板); 之后72 ℃延伸10 min, 最后从65 ℃开始, 以每步0.5 ℃的速度升高至95 ℃, 每个温度保持1 s, 绘制溶解曲线。每个样品3个生物学重复。
| 表1 象草差异表达基因qRT-PCR引物 Table 1 Primers used for expression pattern validations of selected unigenes by qRT-PCR |
用Excel软件初步处理数据, 采用SAS 8.2统计软件进行单因素方差分析。
选择大田种植条件下的象草eg7和eg87进行茎细胞壁组分分析, 结果显示两基因型间茎半纤维素含量无显著差异(P> 0.05), eg7茎中纤维素和酸性洗涤木质素含量比eg87中分别高20.3% (P< 0.05)和186.5% (P< 0.01)(表2)。
| 表2 象草基因型eg7和eg87茎细胞壁组分比较 Table 2 Comparison of cell wall components in stems of genotypes 7 (eg7) and 87 (eg87) (%) |
采用Illumina Hiseq 2000高通量测序技术对象草茎组织进行转录组测序, 共得到了169630902条原始片段, 过滤去除低质量片断和接头后获得153204888条、共13788439920 nt的clean read, 其Q20%为97.25%~98.48%(表3)。原始测序数据已提交到NCBI的序列片段归档(sequence read archive, SRA)数据库(BioprojectID: PRJNA357342)。经Trinity程序(http://trinityrnaseq.sourceforge.net/)对象草转录组clean read进行无参拼接后, 共获得317216 个contigs (eg7:148201; eg87:169015), 其中≥ 500 nt的contig序列有37155个(eg7:17259; eg87:19896)(表4), 在contig数据基础上, 进一步对序列进行组装, 共获得87641个unigene序列, 平均长度为580 nt, N50为780 nt, 其中12863 unigene (14.68 %)序列长度大于1000 nt(表5)。
| 表3 测序数据输出质量情况 Table 3 Summary of the sequencing reads for C. purpureus |
| 表4 象草eg7和eg87转录组数据contig组装统计 Table 4 Contig statistics of Ilumina transcriptome assembly for C. purpureus |
| 表5 象草eg7和eg87转录组数据unigene组装统计 Table 5 Unigene statistics of Ilumina transcriptome assembly for C. purpureus |
为了预测象草unigene的功能, 对unigene进行蛋白编码序列(coding sequences, CDS)预测, 比对到蛋白库的CDS有56725个, 理论的CDS有3532个, 预测到CDS的unigene共60257个(68.75%)。将unigene核酸序列及预测的蛋白序列分别与NCBI Nr、NCBI Nt、Swiss-Prot、GO、COG和KEGG数据库进行比对, 通过序列相似性对unigene进行功能注释, 最终87641条unigenes中有62557条(71.38%)获得注释, 其中Nr和Nt数据库注释的信息最多, 分别是64.35%和63.33%。统计结果见表6。
| 表6 象草unigene在6个数据库中的功能注释情况 Table 6 Summary of functional annotation of assembled unigenes from C. purpureus |
近缘物种相似序列匹配分析结果见图1, 与高粱(Sorghum bicolor)数据库中序列相似的象草unigene最多, 比例高达44.8%, 其次为玉米(Zea mays, 29.8%)、粳稻(Oryza sativa subsp. Japonica, 9.2%)、短柄草(Brachypodium distachyon, 3.6%)、籼稻(Oryza sativa subsp. Indica, 2.6%)以及大麦(Hordeum vulgare, 1.7%)。
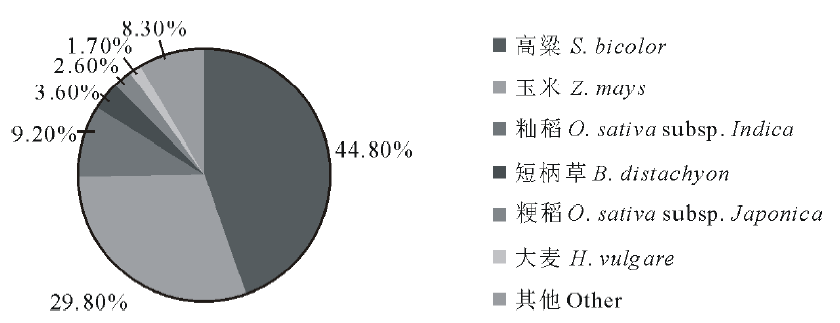 | 图1 Nr注释的物种分布Fig.1 Annotation of species distribution from Nr database |
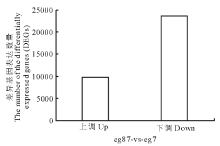
对eg87(对照)与eg7(处理)进行差异基因(differentially expressed genes, DEGs)筛选, 经筛选, 共鉴定出33323个差异表达的基因, 其中上调基因有9704个, 占差异表达基因总数的29.12%; 下调基因共23619个, 占差异表达基因总数的70.88%, 结果如图2所示。
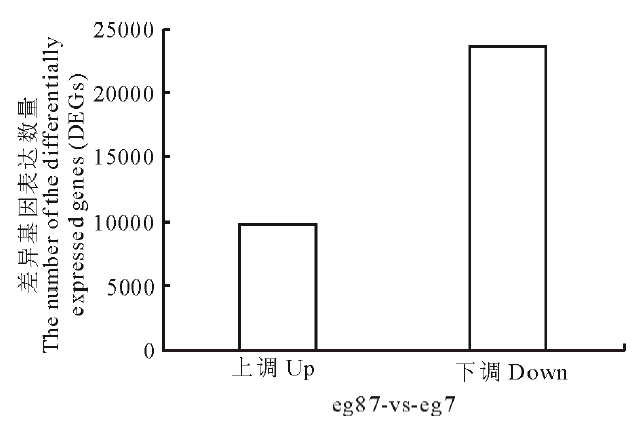 | 图2 象草eg7和eg87差异表达基因Fig.2 The distribution of differentially expressed genes (DEGs) |
GO分析是一种常用的基因功能分析方法, 它按照生物途径(biology process), 细胞组分(cellular component)和分子功能(molecular function)3个大类将基因分别归入一个个功能类群, 并对基因进行注释; 代谢通路(pathway)分析则是将基因具体到代谢网络和代谢通路的特定位置。GO分析和pathway分析有助于我们了解差异基因主要富集在哪些功能类群, 导致哪些代谢通路发生显著改变, 这些信息对于机制研究显得尤为重要。
对eg7与对照组eg87之间的差异表达基因进行GO功能分析发现:所有差异基因被归为54个功能类别, 统计每一功能类群差异基因数目, 其中细胞过程(cellular process)、代谢过程(metabolic process)、细胞(cell), 细胞部分(cell part)、捆绑(binding)、催化活性(catalytic activity)等变化最为显著(图3)。
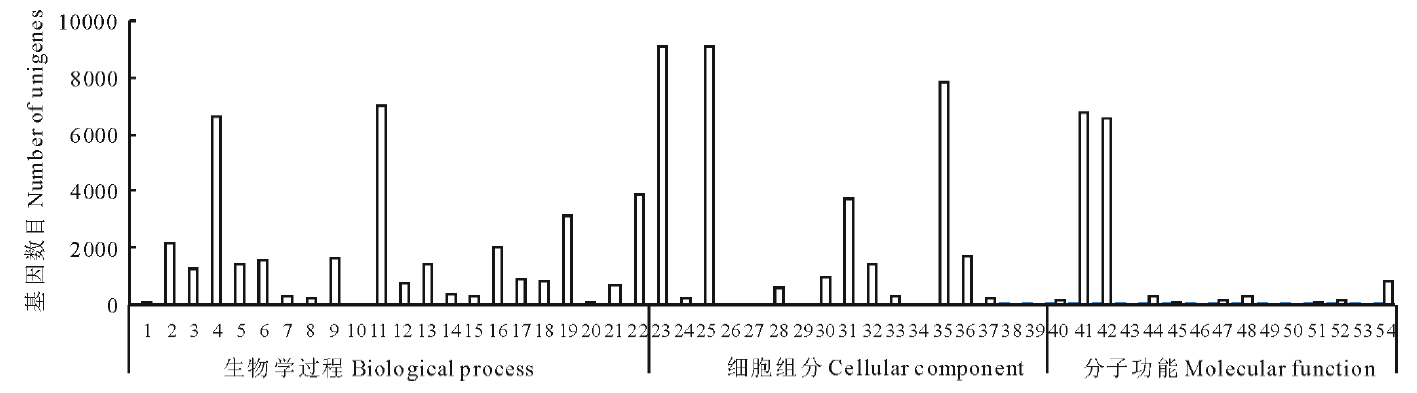 | 图3 象草eg7和eg87(CK)差异表达基因GO富集分类Fig.3 GO enrichment classification of differentially expressed genes 1:生物粘附Biological adhesion; 2:生物调节Biological regulation; 3:细胞成分组织或生物发生Cellular component organization or biogenesis; 4:细胞过程Cellular process; 5:发育过程Developmental process; 6:定位建成Establishment of localization; 7:生长Growth; 8:免疫系统过程Immune system process; 9:定位Localization; 10:运动Locomotion; 11:代谢过程Metabolic process; 12:多生物过程Multi-organism process; 13:多细胞生物过程Multicellular organismal process; 14:生物过程负调控Negative regulation of biological process; 15:生物过程正调控Positive regulation of biological process; 16:生物过程调节Regulation of biological process; 17:繁殖Reproduction; 18:繁殖过程Reproductive process; 19:刺激响应Response to stimulus; 20:节律过程Rhythmic process; 21:信号Signaling; 22:单一生物过程Single-organism process; 23:细胞Cell; 24:细胞连接Cell junction; 25:细胞部分Cell part; 26:细胞外基质Extracellular matrix; 27:细胞外基质部分Extracellular matrix part; 28:细胞外区域Extracellular region; 29:细胞外区域部分Extracellular region part; 30:大分子复合物Macromolecular complex; 31:膜Membrane; 32:膜部分Membrane part; 33:膜封闭的内腔Membrane-enclosed lumen; 34:类核Nucleoid; 35:细胞器Organelle; 36:细胞器部分Organelle part; 37:共质体Symplast; 38:病毒Virion; 39:病毒体部分Virion part; 40:抗氧化活性Antioxidant activity; 41:捆绑Binding; 42:催化活性Catalytic activity; 43:通道调节器活动Channel regulator activity; 44:电子载体活性Electron carrier activity; 45:酶调节剂活性Enzyme regulator activity; 46:金属伴侣活性Metallochaperone activity; 47:分子转导活性Molecular transducer activity; 48:核酸结合转录因子活性Nucleic acid binding transcription factor activity; 49:营养储藏活动Nutrient reservoir activity; 50:蛋白质结合转录因子活性Protein binding transcription factor activity; 51:受体活性Receptor activity; 52:结构分子活性Structural molecule activity; 53:翻译调节器活动Translation regulator activity; 54:转运蛋白活性Transporter activity. |
将获得的87641条unigene与KEGG数据库进行比对, 共有33590(38.32%)条unigene获得注释, 富集得到127条代谢通路。筛选出15条差异基因显著富集的代谢通路, 占差异基因总量的11.07%, 富集程度最显著的5个代谢通路是光合作用蛋白(photosynthesis-antenna proteins)、光合作用(photosynthesis)、betalain生物合成(betalain biosynthesis)、鞘糖脂生物合成-神经节系列(glycosphingolipid biosynthesis-ganglio series)和苯丙烷类代谢途径(phenylpropanoid biosynthesis), 主要涉及能量代谢、多聚糖合成代谢、次生代谢以及萜类和聚酮化合物代谢等。15条通路中, 苯丙烷类代谢途径相关的差异基因数目最多, 达285条, 占2.55%; 其次为苯丙氨酸代谢(157条, 1.40%)、二苯乙烯类和姜酚合成(133条, 1.19%)、氰氨基酸代谢(108条, 0.97%)和光合作用(98条, 0.88%), 其他通路中差异基因占比相对较低(表7)。
| 表7 象草差异基因富集程度排名前15的代谢通路 Table 7 Top 15 enrichment pathways in the stem of C. purpureus |
苯丙烷类代谢途径和与苯丙氨酸代谢途径与细胞壁, 尤其是木质素合成密切相关。将苯丙烷类代谢通路中所有的差异表达基因(285条)全部挑出来, 其中表达上调的有134条, 表达下调的有151条。在苯丙烷类代谢途径中共有83条unigenes注释为木质素单体合成酶基因, 占整个苯丙烷类代谢通路差异基因的29.1%, 其中表达上调的有64条, 占该通路中上调表达差异基因的47.8%, 其中RPKM比值上调 2、4、8、16、32、64 倍以上基因数目分别为25、18、6、6、1和8个; 表达下调的有19条, 占下调表达差异基因的12.6%。筛选出通路中unigene长度≥ 1000 nt、表达差异大于2倍的可能的木质素单体合成酶基因, 其中上调基因26条, 有14个基因表达上调4倍以上, 下调基因仅2条; 我们选择其中表达差异更显著的CL927.Contig7(苯丙氨酸裂解酶, phenylalanine ammonialyase, PAL)、CL4041.Contig5(肉桂酸-4-羟基化酶, cinnamate 4-hydroxylase, C4H)、Unigene32577和CL11870.Contig2(4-香豆酸辅酶A连接酶, 4-coumarate CoA ligase4, CL)、CL2831.Contig3(羟基肉桂酰CoA转移酶, hydroxycinnamoyl CoA transferase, HCT)、CL4302.Contig2(咖啡酰酸辅酶A-O-甲基转移酶, caffeoyl CoA O-methyltransferase, CCoAOMT)、CL1964.Contig1(肉桂酰CoA还原酶, cinnamoyl CoA reductase, CCR)、CL8338.Contig3(阿魏酸5-羟化酶, ferulate 5-hydroxylase, F5H)、CL6560.Contig1(肉桂醇脱氢酶, cinnamyl alcohol dehydrogenase, CAD)作为木质素单体合成酶候选基因待进一步分析。
在285条苯丙烷类代谢差异基因中, 101条unigenes注释为ClassⅢ 型植物过氧化物酶[peroxidase (EC:1.11.1.7), CIII Prxs], 占整个苯丙烷类代谢通路差异基因的35.4%, 其中表达上调的有22条, 占该通路中上调表达差异基因的16.5%, 其中RPKM比值上调2、4、8、16、32、64倍以上基因数目分别为7、7、2、2、2和2个; 表达下调的有79条, 占下调表达差异基因的52.3%, 其中RPKM比值下调2, 4, 8、16、32、64倍以上基因数目分别为24、13、9、9、3和21个, 且其中20条在eg7中表达量极低(raw reads为0)。我们筛选出通路中unigene长度≥ 1000 nt、表达差异大于2倍的CIII Prxs基因(表8), 其中下调基因14条, 有6个基因表达下调8倍以上, 上调基因3条, 上调2~4倍; 这些基因可能在苯丙烷类和木质素代谢过程中发挥重要作用, 我们选择其中差异最为显著的CL843.Contig2、CL438.Contig3、CL438.Contig4、Unigene22891、CL438.Contig5、CL11176.Contig2和CL467.Contig1作为CIII Prxs候选基因待进一步分析。
| 表8 象草中差异表达的ClassⅢ 型植物过氧化物酶基因 Table 8 Expression of ClassⅢ peroxidase genes in the stem of C. purpureus |
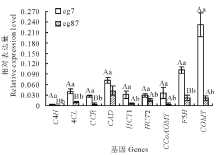
选取参与木质素单体合成的9个差异表达基因, 以Actin和EF1a为内参进行qRT-PCR验证。由图4可知9个木质素单体合成酶基因在eg7的表达量均显著高于eg87, 这与高通量测序结果表达趋势一致。
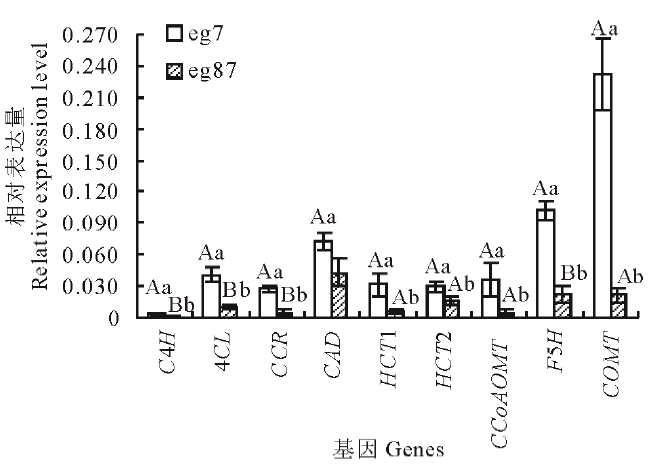 | 图4 差异表达基因的qRT-PCR验证 图中数据表示3次重复的平均值± 标准差; 对eg7和eg87间每一基因相对表达量的差异显著性进行统计分析, 不同小写字母表示差异显著(P< 0.05), 不同大写字母表示差异极显著(P< 0.01)。Fig.4 Validation of selected nine up-regulated transcripts in the eg7 as compared to the eg87 involved in lignin biosynthesis by qRT-PCR Comparison was made between eg7 and eg87, each variable was mean of three time repeated, datas are mean± SD. The different lowercases denoted that the difference was significant (P< 0.05), and the different capitals denoted that the difference was extremely significant (P< 0.01). |
象草是地球上生物产量最高的禾本科、木质纤维素类牧草, 产量高、抗逆性强、用途广泛, 对于盐碱、荒坡等边际性土地的开发利用和重金属、有机物污染土壤的修复利用具有重要应用价值[1, 2, 3, 4, 5]。象草是一种复杂的异源四倍体, 目前尚未完成基因组测序, 没有基因组序列可供参考, 利用无参转录组测序技术挖掘象草关键功能基因具有重要意义。
细胞壁组分占象草干物质重的80%, 细胞壁的主要成分是纤维素、半纤维素和木质素。纤维素和半纤维素是多糖, 在酶的作用下可以转化为可溶性糖供吸收利用, 而木质素能够抵抗自然界多数微生物的有效降解[22], 大量研究表明木质素是限制木质纤维素类作物高效利用的关键因素之一[22, 23]。
在象草eg7与eg87差异表达基因KEGG通路分析中, 光合作用、betalain生物合成、鞘糖脂生物合成-神经节系列和苯丙烷类代谢是差异基因富集程度最显著的代谢通路, 主要涉及能量代谢、多聚糖代谢和次生代谢; 而其中参与木质素合成的苯丙烷类代谢途径具有最为丰富的差异表达基因。木质素是一种复杂的酚类聚合物, 木质素单体合成是木质素合成的关键环节, 一般经苯丙烷途径进行合成[24]。木质素单体合成酶基因主要包括:PAL、C4H、C3H、4CL、COMT、CCoAOMT、F5H、CAD和CCR, 在玉米[25]、拟南芥[26]、杨树(Populus tremuloides)[27]、黑麦草(Lolium perenne)[28]、柳枝稷(Panicum virgatum)[29]等多种植物中的研究均发现:调控这些木质素单体合成酶基因的表达, 均能改变木质素的含量或组成。在象草茎转录中我们挖掘出83条差异表达的木质素单体合成酶基因, 其中64条unigene在木质素含量较高的eg7象草中上调表达。对挑选出的9条木质素单体合成酶基因进行qRT-PCR表达分析, 其在两个象草材料中的表达趋势与高通量测序结果一致。
ClassⅢ 型植物过氧化物酶[peroxidase (EC:1.11.1.7), CIII Prxs]是植物特异性蛋白质, 以多基因家族存在于植物中, 基因名为POX、PRX或PER[30]。CIII Prxs具有很多同工酶, 功能多样, 如参与植物激素代谢、活性氧(reactive oxygen species, ROS)等信号分子代谢、植物抗病反应以及细胞壁的松弛和硬化[31]。在植物生长期间, 细胞的伸长与细胞壁的松弛和硬化(木质化、蜡质化等)紧密相关。这两个过程之间的平衡可以通过CIII Prxs的拮抗活性精确控制。CIII Prxs能够通过在H2O2存在下氧化芳香族细胞壁化合物而使细胞壁硬化, 例如氧化木质素单体形成木质素聚合物[30], 或者通过调节局部H2O2浓度或产生活性氧(ROS)破坏细胞壁聚合物中的共价键而松弛细胞壁[32], 还能通过其生长素氧化酶活性控制细胞伸长[33]。因此, 整个细胞伸长、细胞壁的松弛和硬化都可以由不同的CIII Prx亚型以及精细的转录和转录后调控机制所控制[30]。
前人已在玉米[34]、番茄(Lycopersicon esculentum)[35]、烟草(Nicotiana tabacum)[36]、百日草(Zinnia elegans)[37]等多种植物和组织培养系统中证实CIII Prxs参与木质素合成。De等[34]在玉米中分离出三种编码不同CIII Prxs的cDNA, 命名为ZmPox1, ZmPox2和ZmPox3, 原位杂交表明ZmPox2 mRNA大量积累在幼根伸长区木质化的维管组织和表皮中。过表达番茄过氧化物酶基因(TPX1), 转基因植株中木质素含量迅速增加[35]; 反向抑制烟草NtPrx60基因的表达, 烟草中木质素含量显著降低[36]。研究者在百日草悬浮细胞中成功纯化和克隆了过氧化物酶(ZePrx), 研究表明ZePrx在百日草木质部木质化中发挥重要作用[37]。植物的木质部分化受到严格的激素调节, 启动子分析表明ZePrx含有直接响应生长素、细胞分裂素、赤霉素的顺式元件, 同时还含有响应转录因子NAC, MYB, AP2, MADS和Class III HD Zip的靶序列[38, 39]。在木质部分化过程中, 生长素和细胞分裂素通过上调这些转录因子而间接作用或直接作用于ZePrx, 诱导ZePrx表达, 这种效应与生长素和细胞分裂素诱导木质部分化一致[38]。ZePrx直接受GA3调节, 其活性被GA3抑制, 这种效应与GA抑制幼苗下胚轴细胞壁木质化相一致[39]。Herrero等[40]利用拟南芥基因组进行生物信息学分析和数据挖掘, 寻找拟南芥中与ZePrx功能相似的基因。研究者首先通过序列同源性分析, 寻找到9个与ZePrx序列高度同源的序列(AtPrx 4, 5, 52, 68, 67, 36, 14, 49和72); 通过分析其他参数, 如表面电荷、翻译后修饰、氨基酸的位置、稳定性和寿命/半衰期后, 进一步明确AtPrx4, 52, 49和72为过氧化物酶候选基因; 通过对这4个AtPrx进行更深入的启动子序列分析, 发现AtPrx52还包含与ZePrx高度相似的顺式调控元件, 最终确定AtPrx52是最类似于ZePrx的过氧化物酶。后续的试验证据表明AtPrx52确实与ZePrx功能相似, 参与了木质素合成。
8月取样的象草正处于旺盛生长期, 一方面大量茎干细胞细胞壁松弛, 细胞伸长生长, 一方面大量木质素合成, 细胞壁发生硬化, 整个茎秆细胞处于细胞壁松弛和硬化的动态过程中。通过对木质素含量差异显著的象草eg7和eg87进行转录组比较分析, 我们在差异基因高度富集的苯丙烷类代谢途径中找到了101条差异表达的CIII Prxs, 占整个苯丙烷类代谢通路差异基因的35.4%, 其中在木质素含量较高的eg7中22条CIII Prxs表达上调, 79条CIII Prxs表达下调, 这表明CIII Prxs在象草茎干生长、苯丙烷类和木质素代谢过程中发挥重要而复杂的作用。我们选择差异最为显著的CL843.Contig2、CL438.Contig3、CL438.Contig4、Unigene22891、CL438.Contig5、CL11176.Contig2和CL467.Contig1作为CIII Prxs候选基因, 有待进一步开展深入的生物信息学分析和实验分析以明确其真正的功能。
本研究应用第二代高通量测序技术对象草茎组织进行转录组比较测序, 总获得13788439920 nt数据, 平均长度为580 nt的unigene 87641条, 经过蛋白质编码框预测、序列注释和同源性分析, 共62557条unigene获得注释, 象草与高粱序列同源性最高。共鉴定出33323个差异表达基因, 9704个上调表达, 23619个下调表达。KEGG pathway分析显示参与能量代谢、多聚糖代谢和次生代谢的光合作用代谢通路、betalain生物合成通路、苯丙烷类代谢通路差异基因富集程度最高; 参与木质素合成的苯丙烷类代谢通路具有最多的差异基因数目; PAL、C4H、C3H、4CL、COMT、CCoAOMT等木质素单体合成酶基因和ClassⅢ peroxidase酶基因可作为候选基因, 以期通过进一步的生物信息学分析和功能分析揭示调控象草木质素合成的分子机制。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|