

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
沙打旺EST-SSR分子标记开发及其遗传多样性分析
[宫文龙1  , 王赞
, 王赞2 , 赵桂琴1, * , 马琳2 , 韦宝2 , 龚攀2 , 刘希强2 ]
, 王赞, 马琳|
|


作者简介:宫文龙(1994-), 男, 蒙古族,内蒙古赤峰人, 在读硕士。E-mail: 872471822@qq.com
沙打旺是一种高产优质、抗逆性强的多年生异花授粉豆科牧草,但分子标记的缺乏限制了其在遗传育种等方面的研究和利用。本研究旨在开发大量沙打旺EST-SSR分子标记,为沙打旺种质改良和遗传多样性分析提供参考资源。首先利用 De novo转录组测序技术对两个沙打旺种质(CF019650, CF020070)进行RNA-seq测序,并对测序数据进行拼接获得总长度为190587631 bp的151516个unigenes。进一步在其中的30262个unigenes中检测到39163个EST-SSR位点,SSRs分布频率为25.85%。其中6635 (21.93%)条unigenes含有两个及以上SSR位点,复合SSRs有3514个(11.61%)。对所有EST-SSR位点进行引物设计,共得到22367对特异性引物。利用两个沙打旺种质(CF019650, CF020070)对随机合成的100对引物进行初步筛选,其中90对可扩增出目的特异性条带。随机选择其中51对引物对27个沙打旺种质的遗传多样性进行评估,结果表明:51对引物的平均等位基因数、平均多态性信息含量(PIC)、平均期望杂合度(He)和平均观测杂合度(Ho)分别为8.750、0.682、0.719和0.730。主成分及聚类分析结果揭示不同生态型(匍匐或直立)沙打旺种质的遗传分布具有明显的种质特异性,且聚类结果与其地理来源之间具有较高的相关性。新开发的EST-SSR分子标记可促进沙打旺遗传改良和基因组学研究,有助于沙打旺分子标记辅助育种、QTL定位和遗传变异分析。
Erect milk vetch ( Astragalus adsurgens) is a perennial cross-pollinated legume forage with superior yield, high quality, and strong stress resistance. The lack of molecular markers has limited research on, and genetic breeding of, this species. The aim of this study was to develop a large set of expressed sequence tag-simple sequence repeat (EST-SSR) molecular markers to provide reference resources for the improvement and genetic diversity analysis of erect milk vetch accessions. First, RNA-seq sequencing of two erect milk vetch accessions (CF019650, CF020070) was performed by de novo transcriptome sequencing technology, and 151516 unigenes with a total length of 190587631 bp were obtained by splicing the sequencing data. A total of 39163 EST-SSR loci were detected from 30262 unigene sequences at a frequency of 25.85%, of which 6635 (21.93%) contained two or more SSRs, and 3514 (11.61%) were compound SSRs. Primer pairs (PPs) were designed for all EST-SSR loci (in total, 22367 EST-SSR PPs). In addition, 100 PPs were synthesized randomly and preliminarily screened in two accessions (CF019650, CF020070), and 90 of them were determined to be clear and stable EST-SSR markers. Fifty-one PPs were randomly selected to assess the genetic diversity of 27 erect milk vetch accessions. The average allele number, average polymorphism information content, average expected heterozygosity, and average observed heterozygosity values obtained using the 51 PPs were 8.750, 0.682, 0.719, and 0.730, respectively. Principal coordinate and cluster analyses revealed clear germplasm specificity in the genetic distribution of accessions with creeping and erect ecotypes, as well as a relatively high correlation between clusters and geographic origin. These newly developed EST-SSR molecular markers will be useful for the genetic improvement of, and genomic research on, erect milk vetch, and can be used in molecular marker-assisted breeding, quantitative trait loci mapping, and genetic variation analyses.
沙打旺(Astragalus adsurgens)原产于黄河故道地区, 是一种多年生二倍体(2n=2x=16)异花授粉豆科牧草。常生长于阳坡和林缘灌丛中, 主要分布在中国、前苏联、蒙古和北美等地[1]。因其优异的营养价值、较高的生物量和良好的适口性, 现已成为我国东北、华北和西北等地区广泛种植的牧草作物[2]。沙打旺还兼有较强的适应性、抗旱性和固沙能力等优良特性, 使其在干旱、半干旱地区防止土壤侵蚀、保护自然环境和生态恢复等方面发挥越来越重要的作用[3, 4]。近年来, 关于沙打旺的研究主要集中在形态学、生理学和细胞学等方面[5, 6, 7, 8]。由于受限于较长的生长周期, 种质资源的遗传改良仍为基于生长习性的表型选择[9]。分子标记的缺乏对沙打旺种质改良和遗传多样性分析产生了很大限制。Huang等[10]利用19个随机扩增多态性DNA(random amplified polymorphic DNA, RAPD)标记和96个简单序列间重复(inter-simple sequence repeat, ISSR)标记对22个沙打旺种质的遗传多样性进行了综合分析, 证明了ISSR作为分析沙打旺种质遗传多样性的分子标记具有比RAPD更高的准确性。李瑞芬等[11]利用RAPD标记对13份不同进化状态沙打旺种质资源的遗传多样性进行了调查研究, 结果表明沙打旺野生材料的遗传多样性均高于育成品种和地方材料。然而, 目前开发的有限沙打旺分子标记仍然难以满足标记辅助育种的要求。因此, 大量开发用于改良沙打旺种质的高度多态性的分子标记是十分必要的。
简单重复序列(simple sequence repeat, SSR), 也称微卫星序列, 是广泛分布于真核生物基因组中的分子标记。按来源不同可将其分为基因组SSR和表达序列标签SSR (EST-SSR), 由转录组测序得到的EST-SSR分子标记具有共显性遗传、多态性高、位点特异性和易检测等优于其他分子标记的多种特点[12, 13], 因此广泛应用于许多物种分子标记的开发和利用。刘欢等[14]通过聚丙烯酰胺凝胶电泳及毛细管法从200对多花黑麦草(Lolium multiflorum) EST-SSR引物中筛选出25对扩增稳定的荧光引物。剡转转等[15]通过白花草木樨(Melilotus albus)转录组数据设计了18182对EST-SSR引物并对所开发的引物进行了筛选, 为草木樨属种质资源的遗传改良及分子辅助育种的研究奠定了基础。传统的EST-SSR标记开发方法不仅效率低, 而且操作复杂且成本较高, 不利于大量分子标记的开发。近年来, 随着测序成本的降低, 转录组测序成为EST-SSR分子标记开发过程中可行且重要的工具[16, 17, 18]。随着基因编码区中大量表达序列标签的测定, 所得到的标记不仅具有基因组SSR的特征, 而且其多态性可能与基因功能直接相关[19, 20, 21]。SSR荧光标记毛细管电泳是一种基于DNA测序仪平台的基因分型方法, 与传统的聚丙烯酰胺凝胶电泳相比, 具有快速、高效、自动化、成本低、灵敏度和准确度高等优势[22], 广泛应用于大量分子标记的开发和利用。
本研究基于转录组测序数据开发和验证不同沙打旺种质中的多态性EST-SSR标记, 并利用这些多态性分子标记对不同沙打旺种质的遗传多样性和遗传关系进行初步研究, 以期为沙打旺育种及种质改良提供良好的分析基础和丰富的参考资源。
本研究使用的27个沙打旺种质均由中国国家牧草种质库(北京)提供(表1)。所有材料均于2017年5月种植在中国农业科学院北京畜牧兽医研究所昌平试验基地。试验采用随机区组设计, 小区面积20 m2(4 m× 5 m), 每小区种植一份材料, 每份材料20株。植株生长6周后每份材料随机选取5个单株幼嫩叶片组织等量混合进行DNA提取, 幼叶基因组DNA采用新型植物基因组DNA提取试剂盒(天根, 北京)根据说明书进行提取。通过1%琼脂糖凝胶电泳检测所提DNA质量, 将所得模板DNA用ddH2O稀释至50 ng· μ L-1, 置于-20 ℃储存备用。
| 表1 供试沙打旺种质 Table 1 List of all the accessions used in this study |
使用两个沙打旺种质(CF019650, CF020070)进行RNA-seq试验(表1)。植株生长6周后收集幼叶和茎的混合样品(每个样品3次重复), 立即置于液氮中并储存于-80 ℃。使用植物总RNA提取试剂盒(天根, 北京)按照说明书进行样品总RNA提取。采用多功能酶标仪(Spectra Max i3, 北京)检测所提RNA浓度, 浓度大于600 ng· μ L-1的样品用于转录组测序。
使用带有Oligo(dT)的磁珠富集mRNA, 在适温下加入打断试剂将mRNA打断成短片段。使用随机六聚体引物以打断后的mRNA为模板合成第一链cDNA。利用缓冲液、dNTP、RNaseH和DNA聚合酶I合成第二链cDNA。采用Min Elute PCR纯化试剂盒(天根, 北京)进行纯化回收、粘性末端修复和3'端加A处理。连接测序接头, 通过1%琼脂糖凝胶电泳选择合适大小的片段进行PCR扩增。最后, 通过壹基因公司(北京)的Illumina HiseqTM 2000测序平台对构建好的cDNA文库进行测序。再通过碱基调用图像数据转化为测序读数后获得原始reads。对reads进行过滤并去除污染序列和含有未知核苷酸比率> 5%的reads从而获得高质量的clean reads。使用默认参数的短序列装配程序Trinity (http://sourceforge.net/projects/trinityrnaseq)进行De novo转录组组装。本研究获得的原始序列数据已上传至中国科学院北京基因组研究所BIG数据中心基因组序列数据库, 登记号为CRA001062。
利用简单重复序列鉴定工具程序MISA (http://www.pgrc.ipk-gatersleben.de/misa)在unigenes数据集中对潜在的SSRs进行检测。鉴定SSRs的标准是含有单、二、三、四、五、和六核苷酸的序列分别至少重复12、6、5、5、4和4次。
使用Batch Primer 3.0软件进行引物设计。引物设计的主要参数为:1)引物长度为18~28 bp, 最佳为23 bp; 2) PCR产物大小为80~160 bp; 3)退火温度为55~65 ℃, 最佳温度为60 ℃; 4) GC含量为45%~55%, 最佳值为50%。本研究所用引物均由天一辉远生物公司(北京)合成。PCR扩增采用20 μ L体系:模板DNA (50 ng· μ L-1) 2.0 μ L, 2× Taq PCR Master Mix (10 mmol· L-1 Tris-HCl, pH 8.3; 50 mmol· L-1 MgCl2; 250 μ mol· L-1 dNTPs; 0.5 U· μ L-1 Taq DNA 聚合酶) 10.0 μ L, 10 μ mol· L-1正反引物各0.5 μ L, ddH2O 7.0 μ L。PCR扩增条件为94 ℃预变性5 min; 94 ℃变性30 s, 55 ℃退火45 s, 72 ℃延伸45 s; 30个循环, 72 ℃延伸10 min。通过1%琼脂糖凝胶电泳检测PCR产物。
随机选择扩增条带清晰、具有多态性位点的引物, 在每对引物5'端添加荧光标记FAM(6-carboxy-flourescein), 引物设计标准为选择各位点引物序列间互不干扰、扩增片段长度不重叠、退火温度相近的位点构建二重PCR。对27份沙打旺种质基因组DNA进行二重PCR扩增, 荧光标记引物由华大基因公司(北京)合成。PCR反应选用25 μ L体系: 10× Taq缓冲液 (100 mmol· L-1 Tris-HCl, pH 8.0; 500 mmol· L-1 KCl; 20 mmol· L-1 MgCl2) 2.5 μ L, 2.5 mmol· L-1 dNTP 2.0 μ L, Extaq (5 U· μ L-1) 0.2 μ L, 10 μ mol· L-1正反引物各0.5 μ L, 模板DNA (50 ng· μ L-1) 1.0 μ L, ddH2O 18.3 μ L。PCR扩增条件为: 95 ℃预变性2 min; 95 ℃变性20 s, 54 ℃退火20 s, 72 ℃延伸30 s; 35个循环, 72 ℃延伸10 min。使用ABI3730xl DNA分析仪(美国) 进行自动荧光检测并分离PCR产物。
使用LIZ500分子内标(size standard)和GeneMarker v4.0软件(http://www.appliedbiosystems.com.cn/)对毛细管电泳产物进行数据收集与图像分析。利用PowerMarker v3.25软件计算等位基因数, 观测杂合度(observed heterozygosity, Ho), 期望杂合度(expected heterozygosity, He)和多态性信息含量(polymorphism information content, PIC)[23]。利用PowerMarker v3.25软件计算27个沙打旺种质间遗传距离并创建UPGMA树形图[23], 使用MEGA 7软件进行树形图绘制[24]。采用GenAlEx 6.1软件进行主成分分析(PCoA)[25]。
经过严格的质控和数据过滤, 利用短序列装配程序Trinity共获得151516个unigenes, 总长度为190587631 bp, 平均长度为1258 bp。进一步从30262个unigenes序列中检测到39163个EST-SSR位点, SSRs分布频率为25.85%。在含有ESTs的30262个unigenes中, 6635 (21.93%)个含有两个及以上SSR位点, 3514个(11.61%)为复合型SSRs (表2)。
| 表2 沙打旺EST-SSR标记开发结果 Table 2 Summary of EST-SSR found in erect milkvetch |
从39163个EST-SSR位点的核苷酸基序长度来看:二核苷酸重复是最丰富的基序类型(38.52%), 其次分别为三核苷酸(30.25%)、单核苷酸(23.04%)、五核苷酸(3.00%)、六核苷酸(2.94%)和四核苷酸(2.24%)(表3)。6个基序长度共包含1055种重复类型, 其中单、二、三、四、五和六核苷酸重复分别有4、12、60、116、327和536种(表3)。
| 表3 沙打旺转录组EST-SSR基序类型及分布特征 Table 3 Distribution of the EST-SSR motifs in transcriptome |
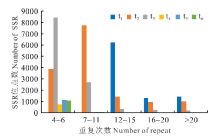
不同EST-SSR基序长度中的重复频率和主要重复基序通常是不同的。本研究中单、二、三、四、五和六核苷酸基序的主要重复频率分别为12~15、6~11、5~8、5~6、4~6和4~6次(图1)。单核苷酸基序类型中最丰富的重复基序是A/T, 其包含95.37%的单核苷酸重复类型, 其次为AG/CT (包含71.39%的二核苷酸重复), AAG/CTT (包含29.18%的三核苷酸重复), AAAT/ATTT (包含28.73%的四核苷酸重复), AAGAG/CTCTT (包含9.19%的五核苷酸重复)和AAGATG/ATCTTC (包含3.73%的六核苷酸重复)(表3)。
 | 图1 SSR重复单元及重复次数的分布Fig.1 The distribution of SSR motifs and repeat number |
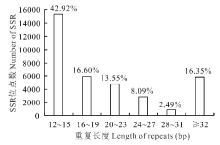
所有沙打旺转录组SSRs序列的平均长度为26.16 bp, 单、二、三、四、五和六核苷酸重复的平均长度分别为19.07、27.13、25.25、27.60、26.20 和31.68 bp。其中分布在12~15 bp的最多, 占总数的42.92%, 其次为16~19 bp (16.60%)、≥ 32 bp (16.35%)、20~23 bp (13.55%)、24~27 bp (8.09%)和28~31 bp (2.49%)(图2)。
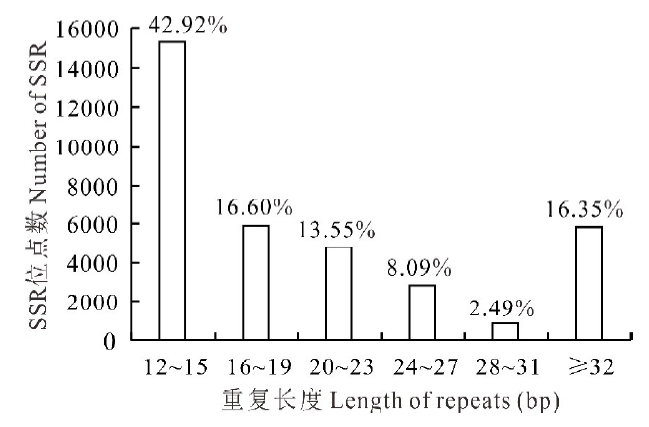 | 图2 SSR重复长度的分布Fig.2 Distribution of the SSR repeat length |
本研究从30262个unigenes中成功设计了22367对引物, 剩余的ESTs由于SSR的侧翼序列太短(< 40个核苷酸)或不符合引物设计的标准从而未能成功进行引物设计。用两个沙打旺种质(CF019650, CF020070)的基因组DNA对随机选择的100对引物(除单核苷酸重复)进行初步筛选, 其中90对引物(90%)可扩增出目的特异性条带, 剩余10对引物产生的扩增条带均不符合预期大小。进一步从成功扩增的90对引物中随机选择51对对27个沙打旺种质基因组DNA进行基因分型(表4和图3)。
| 表4 用于基因分型的引物序列及信息 Table 4 Primer sequences and information for genotyping in this study |
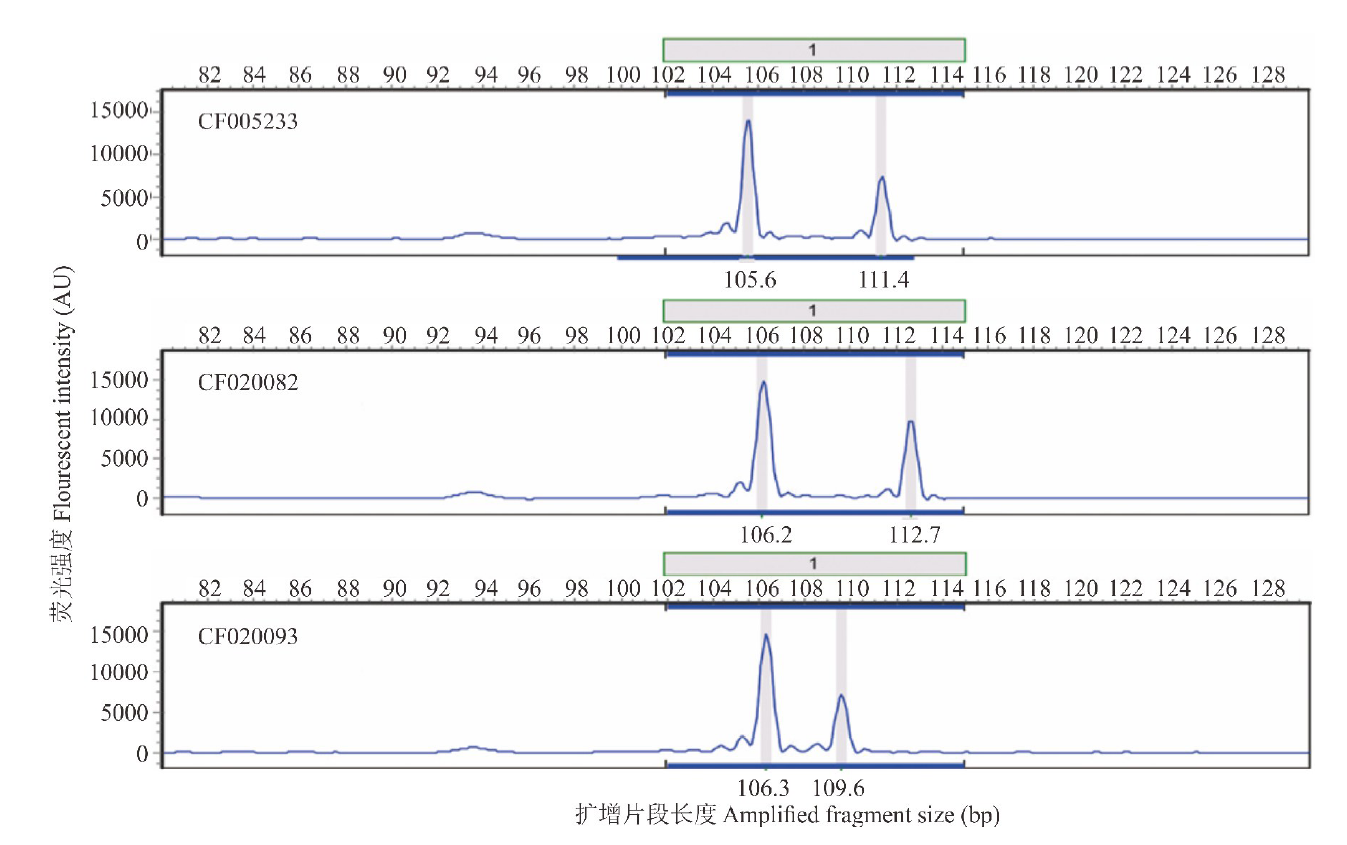 | 图3 3个沙打旺种质在AA01位点的电泳图谱分析Fig.3 Electrophoretogram analysis of one EST-SSR loci (AA01) among 3 erect milkvetch accessions |
基因分型结果显示:51个位点共检测到446个等位基因, 每个EST-SSR位点平均检测到8.75个等位基因, 变化范围从2个(AA26)到17个(AA19)。期望杂合度(He)变化范围为0.235 (AA10)~0.906 (AA38), 平均值为0.719。观测杂合度(Ho)为0.259 (AA10)~1.000 (AA06), 平均值为0.730。多态性信息含量(PIC)为0.224 (AA10)~0.898 (AA38), 平均值为0.682 (表5)。根据Botstein等[26]对多态性的定义, 51个标记中有45个具有高度多态性(PIC> 0.50), 5个具有中度多态性(0.25< PIC< 0.50), 1个标记为低度多态性(0< PIC< 0.25)。
| 表5 51个沙打旺EST-SSR位点遗传多样性 Table 5 Genetic diversity of 51 EST-SSR loci of erect milkvetch |
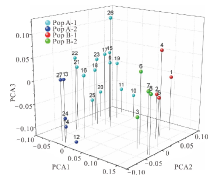
计算27个沙打旺种质间的遗传距离并进行主成分分析(PCoA)(图4), 结果显示前3个主成分分别解释了变异的13.93%, 7.09%和6.35%, 占总变异的27.37%。第一主成分(PCo1)可根据地理分布将27个种质分为两个类群(Pop A和Pop B)。Pop A包括19个种质(13个来自内蒙古, 5个来自西北, 1个来自未知地区), 而Pop B共包括8个种质(6个来自华北, 2个来自东北)。通过第二主成分(PCo2)可将Pop A和Pop B进一步划分为4个子群(Pop A-1, Pop A-2, Pop B-1, Pop B-2)。由图4可知, 这4个子群分别主要由具有直立或匍匐生态型的种质组成:Pop A-1包括14个种质(13个直立种质和1个匍匐种质), Pop A-2包括5个种质(1个直立种质和4个匍匐种质), Pop B-1和Pop B-2分别包括4个直立和4个匍匐种质(图4)。表明地理分布和生态型可以在很大程度上反映沙打旺种质的遗传特征差异。
 | 图4 27个沙打旺种质主成分分析Fig.4 Three-dimensional principal coordinate analysis (PCoA) of 27 erect milkvetch accessions |
为进一步评估沙打旺种质之间的遗传关系, 利用Nei遗传距离构建了27个种质的UPGMA树形图(图5)。UPGMA聚类结果与PCoA分析基本一致。首先确定了两大类群Ⅰ 和 Ⅱ , 结合图4和图5可知, Pop A和Pop B中的种质分别位于聚类组Ⅰ 和聚类组Ⅱ 中。聚类组Ⅰ 可以进一步分为3个亚组, 即Ⅰ -a, Ⅰ -b和Ⅰ -c。Ⅰ -a仅包含1个种质(CF020085)。而来自Pop A-1的大多数种质位于Ⅰ -b (86%)中, 其余种质平均分布在Ⅰ -a (7%)和Ⅰ -c (7%)。Pop A-2的大部分种质都位于Ⅰ -c中(60%), 40%位于Ⅰ -b中 (图5)。聚类组Ⅱ 也可以进一步分为两个亚组, 即Ⅱ -a和 Ⅱ -b。来自Pop B-1的种质全部位于Ⅱ -a中(100%), 而来自Pop B-2的种质则分别位于Ⅱ -a (50%)和Ⅱ -b (50%)中(图5)。聚类结果表明虽然在少数情况下, 来自4个子群的部分种质没有在5个亚组中聚集在一起, 但在大多数情况下大部分种质的聚类结果仍然与其生态型及地理来源具有较高相关性。
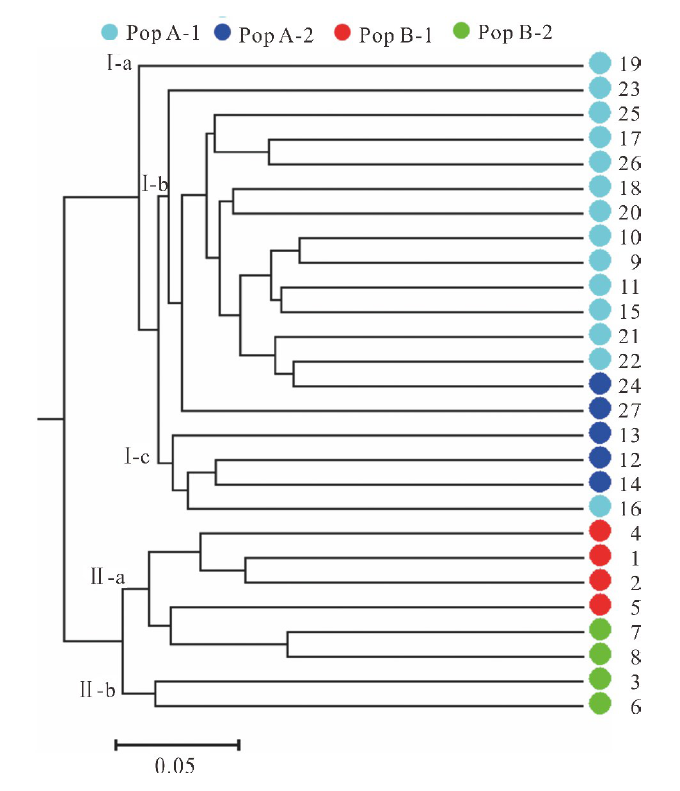 | 图5 27个沙打旺种质UPGMA 聚类分析Fig.5 UPGMA dendrogram of 27 erect milkvetch accessions |
De novo转录组测序已被证明是一种有效且准确的EST-SSR标记开发和鉴定方法, 并已成功应用于许多植物物种中[27, 28]。本研究利用De novo转录组测序从沙打旺的30262条unigenes序列中成功鉴定出39163个EST-SSR位点, SSRs分布频率为25.85%, 高于之前在苜蓿(Medicago sativa)[29]和草木樨(Melilotus)[30]中的报道。SSRs重复基序的类型在不同物种之间通常是不同的, 但在大多数物种中最丰富的重复基序(除单核苷酸重复外)均为二核苷酸和三核苷酸, 这可能与物种的进化历史和基序的基因表达程度有关[31, 32]。本研究中, 最丰富的基序类型同样是二核苷酸重复, 占总重复的38.52%, 这与白菜(Brassica campestris)[33]、珙桐(Davidia involucrata)[34]和橡胶树(Hevea brasiliensis)[35]的研究结果一致。其次最常见的基序类型是三核苷酸重复, 占总重复的30.25%, 也是黄羽扇豆(Lupinus luteus)[36]、老芒麦(Elymus sibiricus)[37]和刺槐(Robinia pseudoacacia)[19]中最丰富的重复基序。此外, AG/CT和AAG/CTT分别是二核苷酸和三核苷酸重复中最常见的重复单元, 这与橡胶树[35]和柠条锦鸡儿(Caragana korshinskii)[38]的研究结果相同。
针对所有EST-SSR位点进行引物设计, 成功获得了22367对特异性引物, 这为沙打旺遗传育种研究提供了丰富的资源。利用两个沙打旺种质基因组DNA对随机选择的100对引物进行初步筛选, 其中90对引物可成功扩增出目的特异性条带, 这一结果高于宁夏枸杞(Lycium barbarum)[18]和苜蓿[29]中的成功扩增比例。较高的扩增率可能是因为本研究中使用的种质均属于具有相似遗传结构的沙打旺种而不涉及其他近缘物种。利用筛选到的多态性引物分析27个沙打旺种质的遗传多样性, 我们在51个EST-SSR位点共检测到446个等位基因, 平均He、Ho和PIC分别为0.719、0.730和0.682, 均高于之前在苜蓿[39]、牛角属(Calotropis)[40]和葱属(Allium)[41]中的报道。上述结果表明本研究中开发的EST-SSR标记具有较高水平的多态性, 适用于沙打旺遗传和育种的研究和应用。此外, 传统上使用聚丙烯酰胺凝胶电泳进行基因分型已被证明是低效且不准确的[42], 本研究中的51个EST-SSR位点均通过自动DNA分析平台进行基因分型, 检测结果更加灵敏准确。
遗传关系分析可以揭示特定种质的遗传多样性, 并可用于标记辅助育种[10]。以前的许多研究都集中在对部分地区沙打旺种质之间的遗传关系进行研究和分析[10, 11], 而本研究包括了来自中国各地区的种质, 结果更加具有代表性和准确性。通过UPGMA聚类分析将沙打旺种质分成两个类群, 其中一个类群包括来自西北和内蒙古的种质, 另一个类群主要包括来自华北和东北的种质, 这可能是由于临近的地理区域和长期的人工选择导致在两个群体中形成了相似的遗传关系。PCoA分析基于直立和匍匐两种生态型将大部分种质(除CF020081和CF008620)划分为4个子群, 这意味着两种不同的生态型可以在很大程度上反映不同沙打旺种质的遗传特征。其余两个种质(CF020081, CF008620)未根据生态型聚集在一起, 可能是因为长期的自然选择导致其遗传背景发生了部分改变。UPGMA聚类结果与PCoA分析结果基本一致, 然而并非所有的种质都基于其地理来源在5个亚组中聚集在一起, 这可能与邻近地区长期的人工选择和种质交换有关。为更全面的了解不同沙打旺种质间的遗传关系, 有必要使用来自中国乃至世界各地的大量不同种质进行遗传多样性及遗传关系评估, 以便为沙打旺种质的改良和利用提供行之有效的手段和重要的参考资源。
本研究通过De novo转录组测序从30262个unigenes中鉴定了39163个EST-SSR位点, 成功设计了22367对引物, 并对100对引物进行验证共筛选出51对多态性引物, 这为沙打旺乃至黄芪属(Astragalus)物种的分子标记开发及利用提供了良好的基础。此外, 对27个沙打旺种质的遗传多样性进行了初步分析, 结果表明本研究开发的多态性EST-SSR分子标记具有较高水平的遗传多样性, 有助于沙打旺分子标记辅助育种, QTL定位和遗传变异研究。主成分和聚类分析表明不同沙打旺种质之间的遗传关系与其地理来源具有较高的相关性, 且不同生态型(直立或匍匐)种质的遗传分布具有明显的种质特异性, 为沙打旺种质改良和遗传多样性研究提供了重要的参考资源。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|