

{kind=link}
{kind=link}
{kind=link}
紫花苜蓿开花性状的遗传特性分析与QTL定位
[何飞1, 2  , 张帆
, 张帆2 , 张铁军2 , 康俊梅2 , 龙瑞才2 , 杨青川2 , 马春晖1, * ]
, 张帆]
|
|


作者简介:何飞(1995-), 男, 新疆阿克苏人, 在读硕士。E-mail: hefei0609@126.com
通过构建紫花苜蓿杂交F1代遗传分离群体,研究紫花苜蓿早熟性状的遗传特性,确定始花期性状的最适遗传模型,同时定位始花期相关的QTL位点。以低产早熟紫花苜蓿(父本)和高产晚熟紫花苜蓿(母本)为亲本构建杂交群体,以亲本和二者杂交产生的152个F1代单株为研究对象。于2015和2016年调查始花期性状,运用主多基因遗传模型分析始花期性状的最适遗传模型。通过GBS测序技术对154个单株进行基因分型,利用测序产生的SNP标记构建连锁图谱,同时结合表型数据进行QTL定位。结果表明:2MG-A为始花期性状的最适遗传模型,2015年的主基因遗传率为99%,2016年的主基因遗传率为98.5%。父本连锁图覆盖图距为1386 cM,平均标记密度3.2 cM;母本连锁图覆盖图距为798.73 cM,平均标记密度8.07 cM。对两年的数据进行QTL定位分析得到2个主效QTL位点,表型贡献率分别为12.1334%和11.0157%。表明始花期主要受两对主效基因控制,同时具有加性作用。始花期性状主要由2个QTL位点控制。
, ZHANG Fan
In order to explore the genetic basis for flowering date variation in alfalfa, a QTL mapping population was set up comprising 152 F1 hybrids of 2 parent plants with contrasting maturity dates, and the QTL loci associated with flowering date were identified. A low-yielding early-maturing alfalfa variety was used as the pollen parent and a high-yield late-maturing variety was used as the seed parent. In 2015 and 2016, the date of initial flowering was recorded for the 152 F1 hybrids and the 2 parent plants, and genotyping of the 154 single plants was carried out using GBS sequencing technology. The SNP markers generated were used to construct a linkage map, and QTL ici Mapping software and phenotypic data were used for QTL positioning of flowering time. A major gene and poly gene inheritance model was then used to build up the optimal genetic model for control of initial flowering time. The analysis indicated two major gene model-additive effect as the optimum genetic model for controlling date of initial flowering. The major gene heritability reached 99.0% in 2015, while it was 98.5% in 2016. The pollen-parent linkage map comprised 1386 cM and the average marker spacing was 3.2 cM. The female-parent linkage map covered 798.73 cM with an average marker spacing of 8.07 cM. Two main QTLs were found in both year’s data sets, with phenotypic variation accounted for being 12.13% and 11.02% respectively. In summary, date of initial flowering is largely determined by two major genes with additive effects. The initial time of flowering is mainly controlled by two QTL sites.
开花性状是紫花苜蓿(Medicago sativa)重要的农艺性状, 产量积累主要发生在开花之前, 通过推迟始花期能够提高产量[1]。开花是植物由营养生长向生殖生长转变的重要标志之一, 对植物的有性生殖起着决定性的作用[2]。始花期与苜蓿干草的产量和品质密切相关, 一直是苜蓿遗传育种过程中的主要关注指标之一。掌握始花期遗传规律能够帮助管理者选择最适合的放牧时期和收获期, 从而获得更高的经济效益[3]。与其他作物相比, 苜蓿开花性状的相关报道比较少, 这主要与紫花苜蓿遗传性状的复杂性及相关分析技术的限制有关。苜蓿为无限花序, 一般群体的开花时间可以延续40~60 d。将始花期作为一个苜蓿开花过程的基准, 有助于更加精细地描述、分析和预测植物的发育。始花期属于多基因控制的数量性状, 由遗传因素和环境因素共同决定[4]。研究始花期性状的遗传构成对深入研究开花性状的遗传规律具有重要意义。前人利用紫花苜蓿杂交后代研究不同农艺性状之间的关系已有报道, 通过调控始花期能够获得更高的产量[5]。将始花期具有差异的材料进行杂交能够获得始花期性状差异明显的F1代材料[6]。但是从表型性状进行紫花苜蓿早熟遗传的相关研究国内还未见报道, 而通过IECM算法计算主+多基因混合遗传模型是分析数量性状遗传规律的有效方法[7]。在紫花苜蓿研究中, 利用主多基因进行表型性状分析的研究还比较少, 这主要是因为主多基因一般应用于二倍体联合世代分析[8]。但是利用该方法分析F2代遗传模型的研究也有报道[9], 这说明利用主多基因模型进行遗传分析也可以应用于单个世代。曹锡文[10]开发出主多基因SEA软件, 这也为主多基因分析提供了极大的便利条件。
紫花苜蓿为同源四倍体, 其杂合水平高, 导致多种构图群体存在偏差。由于多倍体复杂的遗传特性和相关的辅助构图软件的限制, 使得遗传图谱的构建相对困难。借助二倍体分析软件对紫花苜蓿进行研究是一种有效的方法[11]。分子遗传连锁图谱的构建是数量性状基因定位、分子标记辅助育种以及基因克隆的重要理论基础, 利用遗传连锁图谱寻找分子标记与控制优良性状基因之间的连锁关系, 对植物基础遗传育种研究具有重要的应用价值[12]。王梦颖等[13]对早熟材料产生的杂交F1群体进行分析, 初步构建出紫花苜蓿遗传连锁图谱。贾瑞等[14]也利用15个杂交组合产生的F1代单株进行了生物学性状测定。近年来植物遗传图谱的构建研究相对来说比较多, 北柴胡(Bupleurum chinense)[15]、甘薯(Dioscorea esculenta)[16]等植物的遗传图谱已经被构建。但是利用紫花苜蓿进行遗传连锁图谱研究的报道仍然较少, 使得相关QTL定位分析相对困难, 难以加快苜蓿的育种进程。GBS技术的出现弥补了分子标记数量的限制, 较传统的标记检测方法来说能够获得更多的SNP标记, 提高遗传连锁图谱的密度。本研究利用紫花苜蓿花期差异明显的F1代群体进行相关研究, 并结合GBS测序技术进行遗传分析, 重点分析始花期性状的遗传特性, 并进行早熟性状的QTL定位分析, 以期利用植物遗传连锁图以及QTL定位方法, 为解析紫花苜蓿开花性状的遗传规律、培育高产苜蓿新品种提供理论依据。
供试材料为中苜系列品种选育过程中的中间材料:低产早熟紫花苜蓿(父本, 来源于沧州苜蓿)、高产晚熟紫花苜蓿(母本, 来源于中苜1号 )单株个体及二者杂交产生的152个F1代单株个体。
杂交群体表型观测试验于2014-2016年在中国农业科学院国际农业高新技术产业园基地(河北省廊坊市境内)进行。该地位于河北省西北部, 属暖温带大陆性季风气候, 年平均气温11.9 ℃左右, 最冷月份(1月)平均气温为-4.7 ℃, 最热月份(7月)平均气温为26.2 ℃。年降水量554.9 mm。土质为中壤土, 含有机质1.69%, pH值7.37。
杂交种子在温室培养获得F1代幼苗, 对幼苗进行DNA提取并按照王梦颖等[13]的方法利用SSR分子标记鉴定杂交种, 最终获得152个F1代单株个体。通过扦插获得亲本和F1代单株个体的3个重复。采用随机区组排列进行试验, 设3次重复, 行距1 m, 株距0.8 m。在2014年入冬前进行一次刈割, 从而保证不同单株间的一致性。于2015和2016年进行始花期的调查。每年从5月1日起统计不同单株的开花情况, 始花期计算从连续5 d日平均温度大于10 ℃算起(2015年3月10日; 2016年3月16日), 直至第一朵小花出现即表示开花所需天数。
通过对测序数据进行分析, 获得20334个亲本间具有差异的SNP位点。按照Li等[11]对紫花苜蓿遗传连锁图谱构建过程中的分析, 仅利用亲本间的单杂合位点分析来解决同源四倍体分离复杂的简便算法, 并且具有较高的准确性[17]。本研究筛选出缺失值小于10%的单杂合SNP位点(AAAAXAAAB型), 得到7468个SNP位点(杂合父本5888个, 杂合母本1580个)。最后筛选出具有子代分离比为1∶ 1(P> 0.001)的候选位点1320个, 杂合父本位点1011个, 杂合母本位点309个(表4, 表5)。以这些SNP位点为遗传标记进行连锁图谱构建及QTL定位。
将2015, 2016年的154个单株的3个重复均值进行基本统计分析, 并利用3个重复的亲本数据进行亲本差异性T检验; 运用主多基因遗传模型分析遗传率。利用R绘制正态分布图。根据植物数量性状主多基因混合模型分析方法对不同表型性状进行分析。采用极大似然法和IECM算法估计各世代、各成分分布的参数, 利用AIC值,
由表1结果可知, 两年试验结果具有一致性, 并且试验重现性较好。亲本间始花期数据差异明显, 平均开花时间相差达13 d, T检验表明亲本间差异达到极显著水平, 同时Levene’ s 检验证明亲本间方差具有同质性。F1代均值介于双亲之间, 但是变异系数不大。F1代变异范围超过双亲差异范围, 这表现出超亲分布的特点。
| 表1 亲本和杂交F1代的始花期性状基本统计分析 Table 1 Summary statistics analysis of phenotypes for early flowering-time in the F1 progeny and parents |
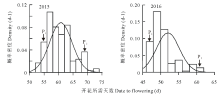
从图1的概率密度分布图中可知, 大部分单株开花时间集中在一个区域, 但是有少部分植株开花时间集中在另一个区域。父本和母本的始花期差异达到极显著水平(P< 0.01)。同时父本开花较早, 母本开花较晚, 亲本差异明显。另外152个F1代植株的开花期差别也很大, 最早开花和最晚开花植株间差异能够达到20 d, 同时两年始花期数据结果具有相似的分离特点。
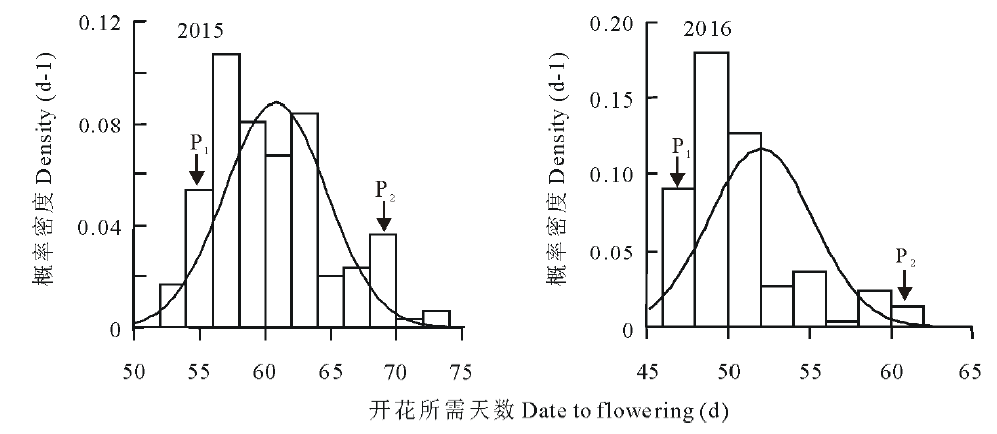 | 图1 2015年和2016年始花期的概率密度分布图 P1, P2分别代表父本, 母本。Fig.1 Density estimate of initial time of flowering in 2015 and 2016 P1, P2 represent paternal and maternal, respectively. |
正态性检验(Kolmogorov-Smirnov)表明, F1代单株分布为非正态分布(P< 0.05), 图1表明F1代单株的分布呈现出双峰分布的特点, 黑色实线为正态分布曲线。
利用数量性状主多基因分析法对F1代单株两年始花期数据进行基因联合分析, 通过极大似然法和IECM算法估算AIC值。根据最适遗传模型筛选原则, 筛选出AIC值最小和较小者, 2015年筛选出2MG-A和2MG-ADI两个最适遗传模型(表2)。并对候选模型进行适合性检验, 结果显示2MG-A的nW2检验达到显著水平, 其他检验未达到显著水平。而2MG-ADI的
| 表2 2015年始花期的遗传参数估计 Table 2 Estimates of genetic parameters for early flowering-time in 2015 |
| 表3 2016年始花期的遗传参数估计 Table 3 Estimates of genetic parameters for early flowering-time in 2016 |
根据不同遗传模型的极大似然估计值, 确定不同遗传模型的遗传参数(表2和表3)。同时根据AIC值和适合性检验得到最适遗传模型为2MG-A和2MG-ADI。2015年试验结果表明, 二者主基因遗传率都达到98%以上, 但是2016年结果仅有2MG-A达到98%, 因此本研究判断候选模型为2MG-A, 主基因的遗传率为99%。始花期主要受两对主效基因控制, 同时具有加性作用。
将上步筛选到的SNP标记(20334个)导入Joinmap软件中, 按照SNP标记所在的染色体进行连锁群设定标准, 以LOD=3为标准构建连锁图谱, 进而以重组率小于0.4和LOD值大于1进行SNP标记的排序和筛选, 最终得到遗传图谱。父本连锁图共有430个SNP标记, 覆盖图距1386 cM, 平均标记间图距3.2 cM, 连锁群长度在122.696~208.514 cM, 其中group 8连锁群最短(表4和图2)。母本连锁图共有99个SNP标记, 覆盖图距798.73 cM, 平均图距8.07 cM, 连锁群长度在54.476~148.826 cM, 其中group 6连锁群最短(表5和图3)。
| 表4 父本连锁群基本信息 Table 4 Paternal chain group basic information |
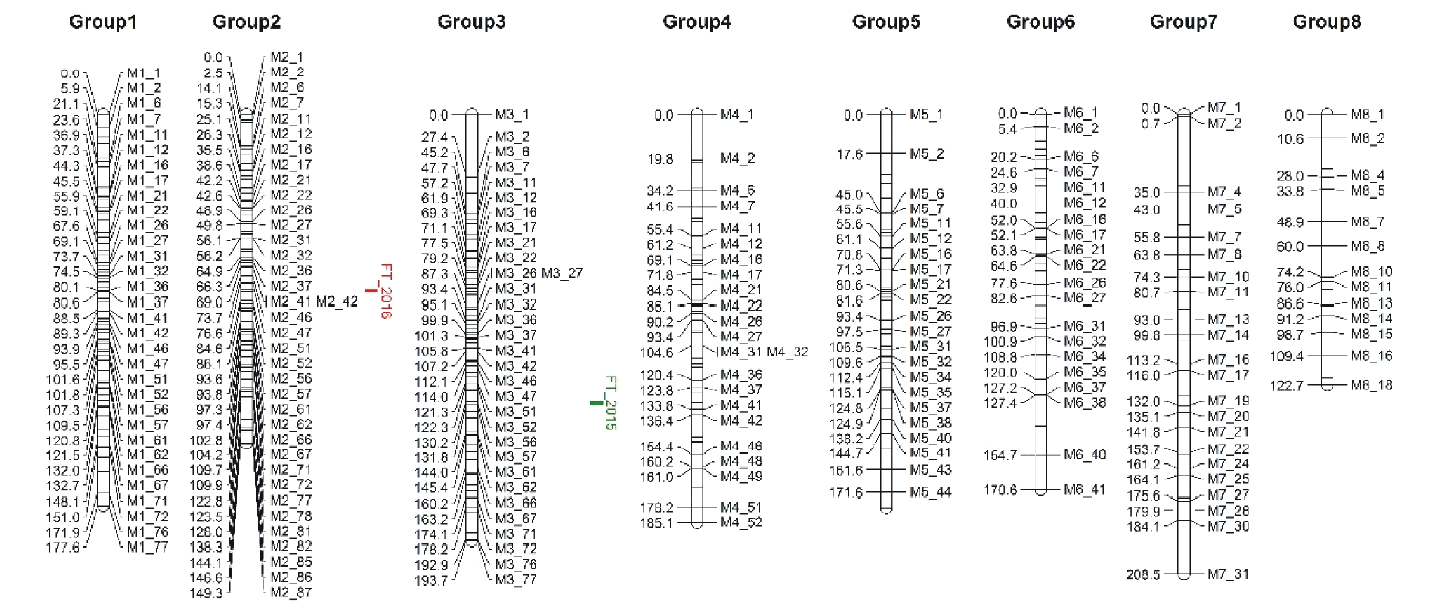 | 图2 父本遗传连锁图 cM表示重组频率的测量单位。左侧表示SNP标记在染色体上的相对位置。右侧代表父本SNP编号。Fig.2 Paternal genetic linkage map cM represents the unit of measurement of the recombination frequency. The left side represents the relative position of the SNP on the chromosome. The right side represents the paternal SNP number. |
| 表5 母本连锁群基本信息 Table 5 Maternal chain group basic information |
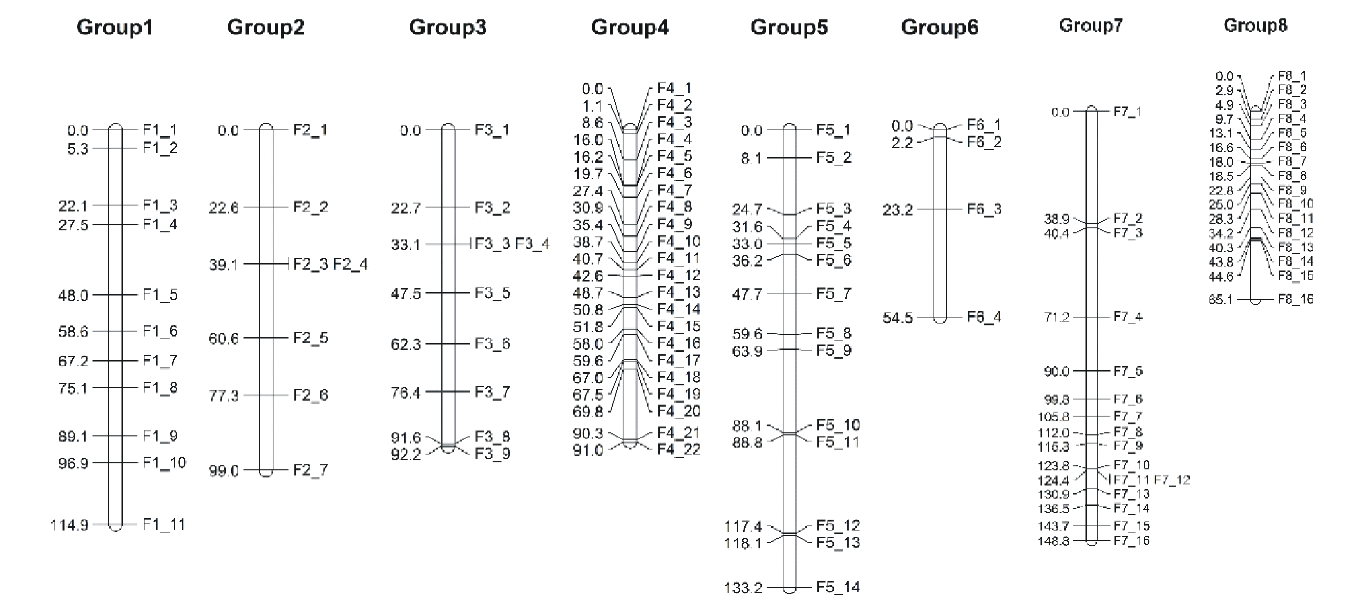 | 图3 母本遗传连锁图 cM表示重组频率的测量单位。左侧表示SNP标记在染色体上的相对位置。右侧代表母本SNP标记的编号。Fig.3 Maternal genetic linkage map cM represents the unit of measurement of the recombination frequency. The left side represents the relative position of the SNP on the chromosome. The right represents the maternal SNP number. |
利用SNP标记信息和2年表型信息进行QTL定位, 最终两年数据分别定位到2个QTL位点。其中2015年结果定位到的QTL位点位于3号连锁群上, 2016年定位到的QTL位点位于2号连锁群上, 两年结果并没有相同的QTL位点(表6)。两个QTL位点的LOD值分别为3.1299 和3.6756, 贡献率分别为12.1334%和11.0157%。
| 表6 父本QTL定位结果 Table 6 Paternal QTL mapping results |
主多基因模型是用传统方法预测表型变异信息的一种有效方法, 它在许多植物中都有应用。周清元等[18]采用主+多基因混合模型预测甘蓝型油菜(Brassica napus)6世代变异信息, 得出果身长、角果长、果喙长等性状的遗传模型, 并根据不同遗传模型的参数结果估计出加性效应和显性效应值, 进而从不同模型间的相互作用中推断出遗传进化过程。李忠南等[19]对玉米(Zea mays)6世代进行联合分析得出, 叶绿素SPAD值主要受两对主基因+多基因控制。同时该遗传模型也可以应用于动物研究[20], 这些模型适合性检验结果良好, 因此该方法非常适合应用于遗传分析研究。同时该方法已经被沿用多年, 在预测准确性方面有一定的优势, 且各种试验结果良好。因此该方法适合作为关联分析的基础步骤, 通过表型预测初步估计遗传变异信息, 为进一步的遗传分析研究提供参考依据。
两年试验结果, 能够通过不同年份的试验结果信息消除环境因素, 进一步通过不同环境中稳定存在的主多基因模型预测始花期性状的遗传变异规律。两年数据结果具有一致性, 始花期性状的最适遗传模型为2MG-A和2MG-ADI, 这两个遗传模型在不同年份分别具有最小的AIC值, 同时主基因遗传率能够解释98%以上的表型变异信息。此外两个遗传模型都是由2对主效基因控制, 因此可推断始花期性状由两对主效基因控制。但是2MG-ADI的主基因遗传率在2016年为0.45%, 因此并不是最优遗传模型。而2MG-A遗传模型在两年结果中都能解释98%的表型变异信息, 因此始花期性状的遗传信息可以被2MG-A遗传模型较好解释, 同时始花期性状受环境影响因素较小, 该性状主要被遗传因素影响, 相关遗传信息能够在不同环境中稳定表达。
紫花苜蓿为同源四倍体, 异花授粉植物, 其杂合水平高, 因此导致多种构图群体存在偏差。由于多倍体复杂的遗传特性和相关的辅助构图软件的限制, 使得遗传图谱的构建相对困难[21]。在四倍体苜蓿中已经建立了一些基因连锁图谱。应用QTL作图, 一些重要的农艺性状, 如产量[22, 23]、耐寒性等[24, 25]QTL已鉴定出来。同时已有大量试验证实在蒺藜苜蓿(Medicago truncatula)中鉴定出的QTL, 对苜蓿的改良具有应用价值。如蒺藜苜蓿的遗传图谱在抗春季黑茎病和叶斑病的QTL中被确定[26], 而且与氮素营养[27, 28]及形态特征[29, 30]相关的QTL也被鉴定出。因此, 蒺藜苜蓿QTL定位与遗传图谱构建的研究为四倍体苜蓿的相关研究奠定了良好的基础[26, 27, 28, 29, 30, 31]。目前只有为数不多的几个完整的紫花苜蓿的遗传图谱。最早的报道是用F1群体, 分析32个RAPD标记的分离结果, 发现了9个连锁群, 并且构建了四倍体苜蓿的遗传图谱[32]。Irwin等[33]利用RAPD和AFLP标记对紫花苜蓿亲本进行抗炭疽病研究, 共得到了10个与抗炭疽显著相关的标记。Musial等[34]运用RAPD、AFLP和SSR技术从紫花苜蓿的遗传图谱中得到与产量正相关的标记及与产量负相关的基因。然而, 大多数分子标记, 如SSR等不能够满足基因的精细定位和全基因组关联研究[17]。SNP标记构建遗传图谱, 具有共显性, 位点稳定等优点, 是构建遗传图谱较为理想的分子标记之一。本试验通过对测序数据进行分析, 获得20334个亲本间具有差异的SNP位点。最后筛选出具有子代分离比为1∶ 1(P> 0.001)的候选位点1320个, 杂合父本位点1011个, 杂合母本位点309个, 最终构建了包含8个连锁群的遗传图谱。父本遗传图谱覆盖图距为1386 cM, 标记间平均图距3.2 cM, 母本覆盖图距798.73 cM, 平均图距8.07 cM。同时定位到早熟性状相关的两个QTL位点。本结论和前人研究结果存在一定差异[11], 这主要是分析方法的差别。本研究利用蒺藜苜蓿作为参考基因组进行SNP分型, 这样进行分析的优点是SNP在染色体上的位置能够确定, 但是定位到染色体上的SNP标记较少。结合性状鉴定结果, 本研究定位到早熟性状QTL位点, 并且表型解释率较高, 因此可以说明本研究的分析方法具有现实可行性。同时也说明这两个标记附近是控制开花性状的重要区域, 这些区域的发现为紫花苜蓿早熟育种的分子标记辅助选择提供了有利证据。
1)利用数量性状主多基因分析法对紫花苜蓿杂交F1代单株的两年始花期数据进行基因联合分析, 筛选出2MG-A为最适遗传模型。2015年的主基因遗传率为99%, 2016年的主基因遗传率为98.5%。始花期主要受两对主效基因控制, 同时具有加性作用。
2)构建了紫花苜蓿遗传连锁图谱, 其中父本遗传图谱覆盖图距为1386 cM, 标记间平均图距3.2 cM; 母本覆盖图距798.73 cM, 平均图距8.07 cM。QTL定位分析获得2个早熟相关QTL位点, LOD值分别为3.1299 和3.6756, 贡献率分别为12.1334%和11.0157%。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|