


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
不同侵染时间点稻粒黑粉病菌的转录组分析
[舒新月1, 2, **  , 江波
, 江波3, ** , 马丽1, 2 , 郑爱萍1, 2, * ]
, 江波, 马丽]
|
|


作者简介:舒新月(1995-),女,四川雅安人,在读硕士。E-mail: 1374055882@qq.com;江波(1974-),男,重庆涪陵人,副教授,硕士。E-mail: 543038089@qq.com。**共同第一作者These authors contributed equally to this work.
由土传真菌稻粒黑粉菌引起的稻粒黑粉病是水稻杂交制种田中的主要病害之一。为初步解析稻粒黑粉病菌侵染过程中与寄主的互作分子机制,本研究用稻粒黑粉病菌强毒力菌株JY-521侵染感病不育系材料“9311A”,于侵染8、12、24、48和72 h 5个时间点取菌样进行转录组测序分析。结果表明,与未侵染对照组相比,在侵染8 h时,差异表达基因(DEGs)数最多,有337个DEGs;其中上调的DEGs有120个,下调的DEGs有217个。337个DEGs 的KEGG富集分析表明,脂肪酸代谢和过氧化物酶体可能是稻粒黑粉病菌侵染过程中的关键代谢途径。此外,对编码碳水化合物酶、分泌蛋白和宿主植物—病原互作蛋白的DEGs在5个侵染时间点的表达模式进行解析,结果表明,一些显著上调表达的DEGs可能是稻粒黑粉病菌侵染的关键致病因子。该研究结果为解析稻粒黑粉病菌的致病分子机制,进一步防控该病害奠定了理论基础。
Rice kernel smut (RSB) caused by Tilletia horrida, is one of the most important rice diseases in hybrid rice growing areas worldwide. In order to classify the mechanisms of pathogenicity, transcriptome analysis of the T. horrid strain JY-521 was conducted at different times post inoculation (8, 12, 24, 48, and 72 h) early in the infection. The highest number of differentially expressed genes (DEGs) occurred at 8 h post inoculation. Based on kyoto encyclopedia of genes and genomes (KEGG) pathway analysis of the DEGs, autophagy processes and lipid degradation were key pathways for T. horrida pathogenicity. The expression patterns of carbohydrate-active enzyme genes, pathogen-host interaction genes, and secreted protein genes were also analyzed at different times during the infection, and some DEGs that may play an important role in pathogenic progress of T. horrid were found. In summary, this research provides a new foundation for future study of the infection mechanism and control of this important rice disease.
水稻(Oryza sativa)是我国四大粮食作物之一, 在粮食生产中具有十分重要的地位[1], 有2/3的人口以稻米为主食。水稻稻粒黑粉病(rice kernel smut)主要为害水稻不育系, 其病原稻粒黑粉菌(Tilletia horrida)抗逆性强, 主要以厚垣孢子在寄主种子和土壤中越冬[2], 于水稻扬花期侵染水稻不育系花器官[3]。据报道, 自20世纪70年代以来, 随着杂交稻和高产栽培种在农业生产上的广泛推广, 发病率可高达70%~80%, 在亚洲、美洲、非洲及中国各个稻区均有发生, 稻粒黑粉病已经成为杂交稻制繁种的主要病害[4], 特别是杂交稻中的水稻不育系危害更加严重, 且有逐年加重的趋势[5]。目前, 田间生产中, 主要以化学药剂对其进行防控, 仍没有较为完善的防控体系。转录组测序技术成本较低, 精确度高, 已成功应用于寄主— 病原互作机制的解析和功能基因的挖掘[6, 7]。Thatcher等[8]对尖孢镰刀菌(Fusarium oxysporum)侵染苜蓿进行转录组测序分析, 初步筛选到10个可能在镰刀菌侵染中发挥重要作用的候选效应因子; 韩彦卿等[9]通过对稻曲病菌(Ustilaginoidea virens)侵染水稻抗病品种与感病品种的比较转录组分析初步解析了水稻抗稻曲病的分子机制; Xiao等[10]基于转录组测序技术对小麦(Triticum aestivum)赤霉病菌(FusaHum graminearum)侵染小麦抗病品种与感病品种进行研究, 发现病程相关蛋白PR5、PR14, ABC转运蛋白编码基因上调表达, 其可能在抗小麦赤霉病生物学过程中发挥关键作用, 此外, 茉莉酸(jasmonic acid, JA)途径也参与了对小麦赤霉病的抗病过程。
目前, 对稻粒黑粉病菌的研究大多基于其形态学和进化分析, 关于其致病分子机制的解析报道较少。本研究对稻粒黑粉病菌侵染感病不育系材料9311A分别在8、12、24、48和72 h进行取样, 以未侵染8 h样本作为对照, 共6个样本进行转录组测序分析, 初步解析了稻粒黑粉病菌侵染早期与寄主的可能互作机制, 为深入挖掘稻粒黑粉病菌关键致病因子奠定了理论基础。
稻粒黑粉病菌强毒力菌株JY-521由四川农业大学植物病理实验室分离并保存。高感稻粒黑粉病水稻不育系材料9311A种子由湖北省农业科学院粮食作物研究所提供。
马铃薯蔗糖琼脂(potato-sucrose-agar, PSA)固体培养基:马铃薯200.0 g, 蔗糖20.0 g, 琼脂20.0 g, 蒸馏水1 L, pH 7.0, 120 ℃高压灭菌15 min。
马铃薯蔗糖液体培养基(potato-sucrose-broth, PSB):马铃薯200.0 g, 蔗糖20.0 g, 蒸馏水1 L, pH 7.0, 120 ℃高压灭菌15 min。
1.3.1 样品制备 将稻粒黑粉病菌强毒力菌株JY-521于PSA固体培养基上活化后, 挑取单菌落于新的PSA固体培养基中, 于28 ℃恒温箱中培养3 d。水稻扬花期采集感病不育系9311A幼穗, 75%乙醇消毒2 min, 用无菌水清洗掉多余的乙醇; 杂交剪剪去颖壳一端, 使花器官露出, 放置于稻粒黑粉病菌菌落上。分别于侵染8、12、24、48和72 h刮取颖壳表面菌丝, 以不侵染菌落作为对照(8 h), 用于RNA提取。
1.3.2 样品RNA提取 采用Tri-zol法[11]提取RNA, 用1.0%变性琼脂糖凝胶检测总RNA, 并保存于-80 ℃冰箱备用。
1.3.3 测序分析 利用Illumina Hiseq 2500平台进行测序, 测序模式为125PE。以本课题组完成的稻粒黑粉病菌JY-521全基因组测序作为参考基因组[12]。使用SOAP 2软件将测序得到的过滤后读段(clean reads)比对到参考基因组。表达定量以每千个碱基的转录每百万映射读取的片段数(fragments per kilobase of exon model per million mapped fragments, FPKM)为单位, 参照Benjamini等[13]调控倍性变化率(fold charge ratio, FC)的方法对P值结果进行调整。
1.3.4 差异基因表达模式分析 基于比较转录组分析, 从5个时间点的稻粒黑粉病菌中筛选到500个显著差异表达基因(different expression genes, DEGs), 根据500个基因在稻粒黑粉病菌中的表达量变化, 本研究使用Short Time-series Expression Miner (STEM)软件将其划分为24个聚类群[15]。
1.3.5 KEGG和GO富集分析 使用R程序中的GOseq程序包对DEGs的GO功能进行富集分析[16]; DEGs的KEGG分析使用KOBAS软件[17]。
采用Benjamini等[14]的方法对DEGs进行显著性分析, 以P< 0.05, |log2fold charge|> 1为显著表达差异。KEGG和GO富集的P-value经过Bonferroni校正后, 以Corrected P-value< 0.05为显著富集。
6个样本的转录组测序共获得202624114条clean reads, 去掉接头、含N的序列和低质量的reads后, 高质量clean reads的数据量约为25.1 Gb; 在参考基因组上的比对率达70.16%~73.70%, Q20介于96.51%~96.86%, GC含量为53.82%~54.85%。根据测序的clean reads和比对到参考基因组的数据, 可进行后续数据分析(表1)。
| 表1 稻粒黑粉病菌接种水稻品种9311A在5个不同时间点的转录组测序数据比对到参考基因组JY-521获得的reads Table 1 Statistics of the total reads and reads mapped to the JY-521 reference genome in 9311A rice cultivar infected by T. horrida at five different time points |
与不侵染对照相比, 稻粒黑粉菌侵染8 h时显著上调基因120个, 显著下调基因217个; 12 h显著上调基因62个, 显著下调基因49个; 24 h显著上调基因29个, 显著下调基因62个; 48 h上调显著差异基因64个, 下调显著差异基因25个; 72 h上调显著差异基因32个, 下调显著差异基因23个(图1A)。在上调显著差异基因和下调显著差异基因中, 5个时间点均不存在持续上调和持续下调的基因(图1B, C), 其中大部分显著差异基因集中在稻粒黑粉病菌侵染8 h, 本研究推测8 h可能是稻粒黑粉病菌侵染的关键时间点。
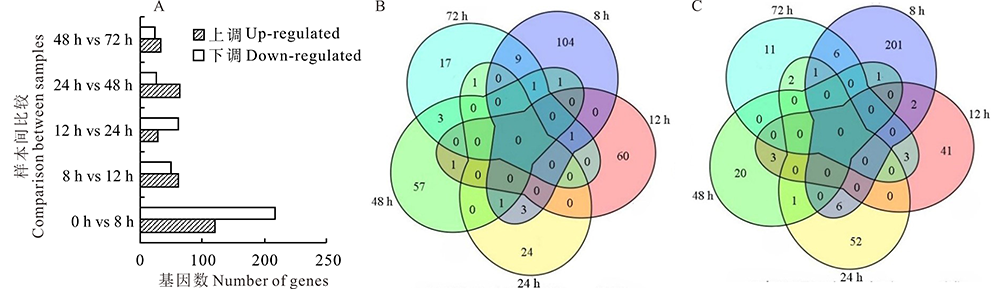 | 图1 稻粒黑粉病菌在5个不同侵染时间点的遗传统计 A: 5个侵染时间点差异表达基因统计Different expression genes statistics at five inoculation time points; B:韦恩图展示5个时间点上调基因的交集Venn diagram of up-regulated genes at five inoculation time points; C:韦恩图展示5个时间点下调基因的交集Venn diagram of down-regulated genes at five inoculation time points.Fig.1 Genetic statistics of five different infection time points of kernel smut |
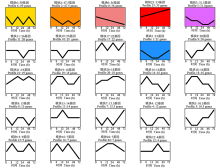
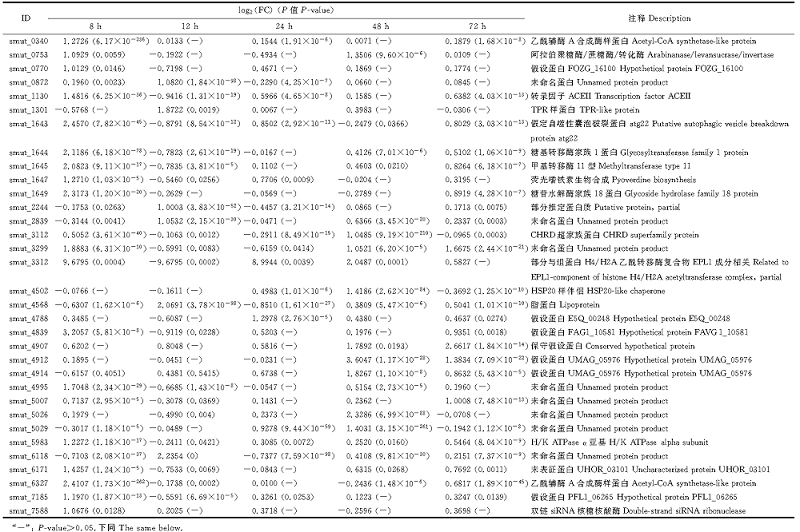
植物真菌的致病反应是由多个基因共同调控的复杂生物学过程[18], 具有相同表达模式的基因可能在同一生物学过程中发挥功能。本研究对5个侵染时间点的500个DEGs进行了聚类分析, 结果表明, 基于其表达模式划分为24个表达模块(图2)。其中在侵染8 h表达量下调的模块(profile6、profile2和profile3)、持续下调表达的模块(profile0)和持续上调表达的模块(profile23)基因数最多。呈持续上调表达模块(profile23)内共有33个DEGs(表2), 包括编码转录因子ACEII、假定自噬性囊泡破裂蛋白atg22、糖基转移酶家族1蛋白、甲基转移酶11型、糖苷水解酶家族18蛋白和CHRD超家族蛋白等与致病性相关蛋白编码基因。呈持续下调表达模块(profile0)内共有36个DEGs(表3), 分别编码假定类固醇5α -还原酶、RTA1样蛋白、多药转运蛋白、 MFS普通基质转运蛋白、短链黄醇脱氢/还原酶SDR、LRR蛋白、小寡肽转运蛋白、WD40重复基序蛋白、甲基转移酶、脱氧核糖二嘧啶光裂合酶、热激蛋白9、谷胱苷肽S-转移酶和MEF2-翻译延伸因子。
 | 图2 稻粒黑粉病菌中共表达差异基因的表达模块 模块顺序根据基因的数量排序。Fig.2 Clusters of genes with unique expression patterns in kernel smut of rice Clusters were ordered according to the number of genes assigned to them. |
| 表2 profile23中持续上调表达基因 Table 2 Sustained up-regulated genes in profile23 |
| 表3 Profile0中持续下调表达基因 Table 3 Sustained down-regulated genes in profile0 |
将筛选阈值定为corrected P-value≤ 0.05, 对不同时间获得的差异表达基因进行GO注释分析(图3), 这些DEGs被富集注释到3个GO分类, 分别是生物学过程(biological process)、细胞组成(cellular component)和分子功能(molecular function)。以未侵染为对照, 对稻粒黑粉菌侵染8 h显著差异表达基因进行GO富集分析, 结果显示, 上调DEGs在生物学过程分类中的生长、细胞组成分类中的细胞器组件和分子功能分类中的酶调节活性、分子功能调节显著富集。显著下调DEGs在生物学过程的代谢过程和分子功能的结合、催化活性显著富集。推测该7条富集条目可能与稻粒黑粉病菌致病途径相关。
 | 图3 稻粒黑粉病菌侵染8 h的DEGs的GO富集分析 1:生长Growth; 2:定位Localization; 3:生殖Reproduction; 4:应激Response to stimulus; 5:单生物过程Single-organism process; 6:信号Signaling; 7:生物调控Biological regulation; 8:代谢过程Metabolic process; 9:细胞成分组织或生物发生Cellular component organization or biogenesis; 10:细胞进程Cellular process; 11:核苷酸结合转录因子活性Nucleic acid binding transcription factor activity; 12:转运器活性Transporter activity; 13:催化活性Catalytic activity; 14:分子传感器Molecular transducer activity; 15:酶调节器活性Enzyme regulator activity; 16:分子功能调节器Molecular function regulator; 17:结合Binding; 18:细胞膜Membrane; 19:细胞膜组件Membrane part; 20:细胞器Organelle; 21:大分子复合物Macromolecular complex; 22:细胞器组件Organelle part; 23:细胞Cell; 24:细胞组件Cell part.Fig.3 DEGs in T. horrida at 8 h of infection by GO enrichment analysis |
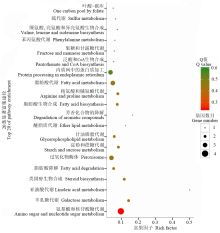
对DEGs进行KEGG富集分析表明, 脂肪酸代谢、氨基糖和核苷酸糖代谢、过氧化物酶体(peroxisome)、脂肪酸降解、类固醇合成、脂肪酸生物合成和淀粉与蔗糖代谢显著富集, 这些代谢途径很可能参与了稻粒黑粉病菌对水稻粒致病过程(图4)。
 | 图4 稻粒黑粉病菌侵染8 h的DEGs显著生物合成途径的KEGG富集分析Fig.4 The enriched biosynthetic pathways of DEGs in T. horrida at 8 h by KEGG enrichment analysis |
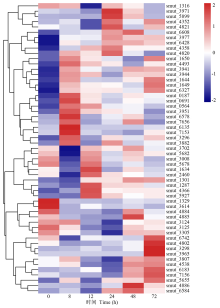
病原性真菌分泌的碳水化合物活性酶可通过降解植物细胞壁使病原菌成功侵染寄主[19]。5个侵染时间点发现55个与碳水化合物酶相关的上调表达DEGs(图5), 其中8 h显著上调DEGs有31个, 12 h显著上调DEGs有20个, 24 h显著上调DEGs有34个, 48 h显著上调DEGs有38个, 72 h显著上调DEGs有39个。其中MFS转运蛋白[major facilitator superfamily (MFS) transporter]编码基因smut_0187和smut_4884、角质酶(cutinase)编码基因smut_3124以及丝氨酸/苏氨酸蛋白激酶(serine/threonine-protein kinase)编码基因smut_4358分别在侵染8、12和48 h上调表达。有研究发现, MFS转运蛋白、角质酶以及丝氨酸/苏氨酸蛋白激酶在真菌致病过程中起关键作用[20, 21, 22], 表明smut_0187、smut_4884、smut_3124和smut_4358可能参与稻粒黑粉病菌致病机制。
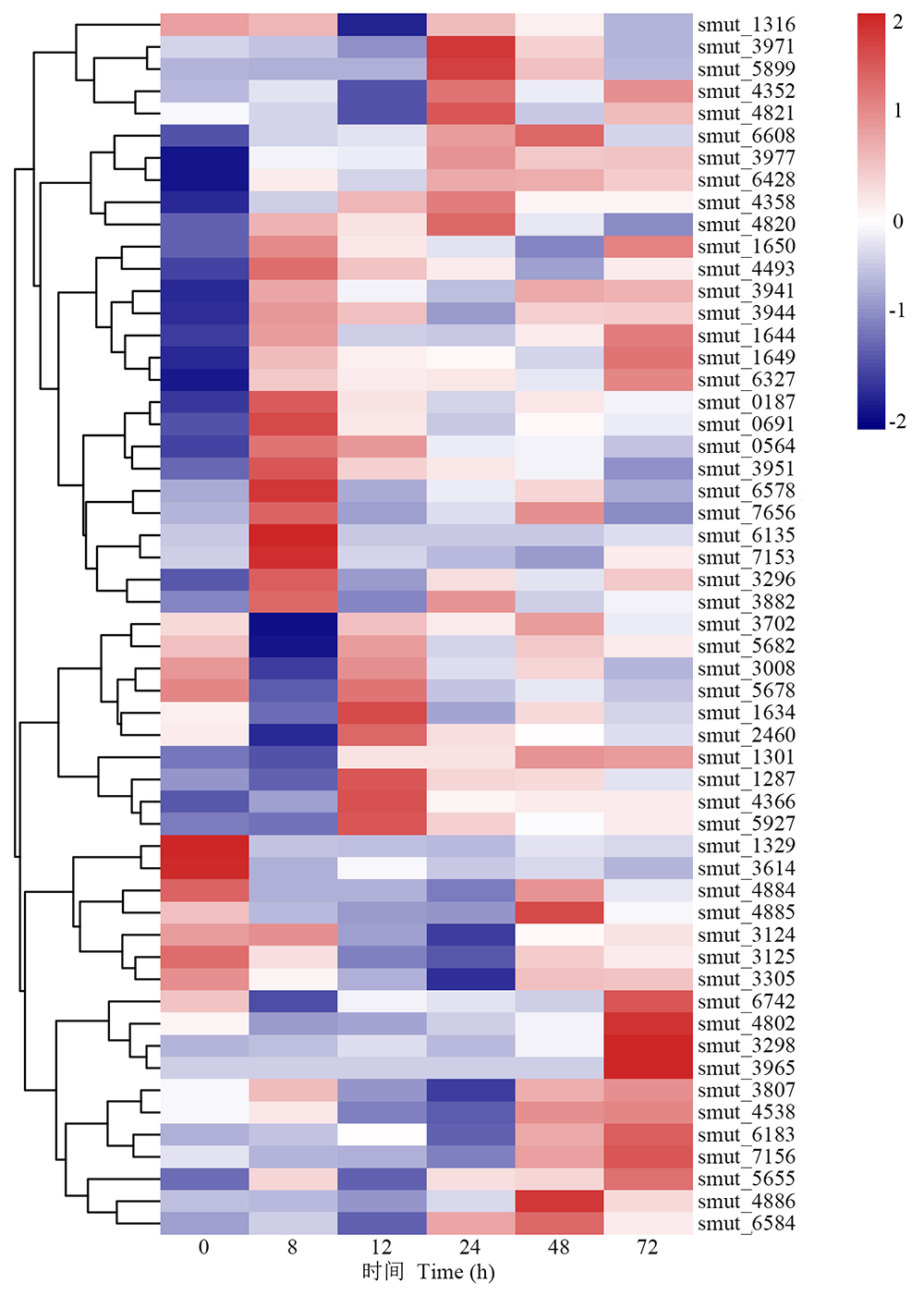 | 图5 病原相关基因在碳水化合物活性酶中的表达模式Fig.5 Expression pattern of pathogenic related genes in carbohydrate-active enzyme |
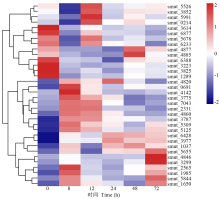
真菌分泌蛋白大多与无毒基因相关, 且在宿主植物的致病性过程中起重要作用。在真菌侵染宿主植物过程中, 分泌蛋白能与宿主植物细胞进行信号交换, 引起一系列生理生化反应[23]。在稻粒黑粉病菌侵染8 h有34个分泌蛋白相关DEGs, 其中20个上调表达, 14个下调表达(图6), 这些基因可能与稻粒黑粉病菌的致病途径相关。其中乙二醛氧化酶(glyoxal oxidase, GLX)编码基因smut_4820在侵染8 h显著上调, 相关报道发现, GLX在真菌侵染宿主细胞时能降解植物细胞壁[24], 表明smut_4820可能参与稻粒黑粉病菌定殖稻穗过程。
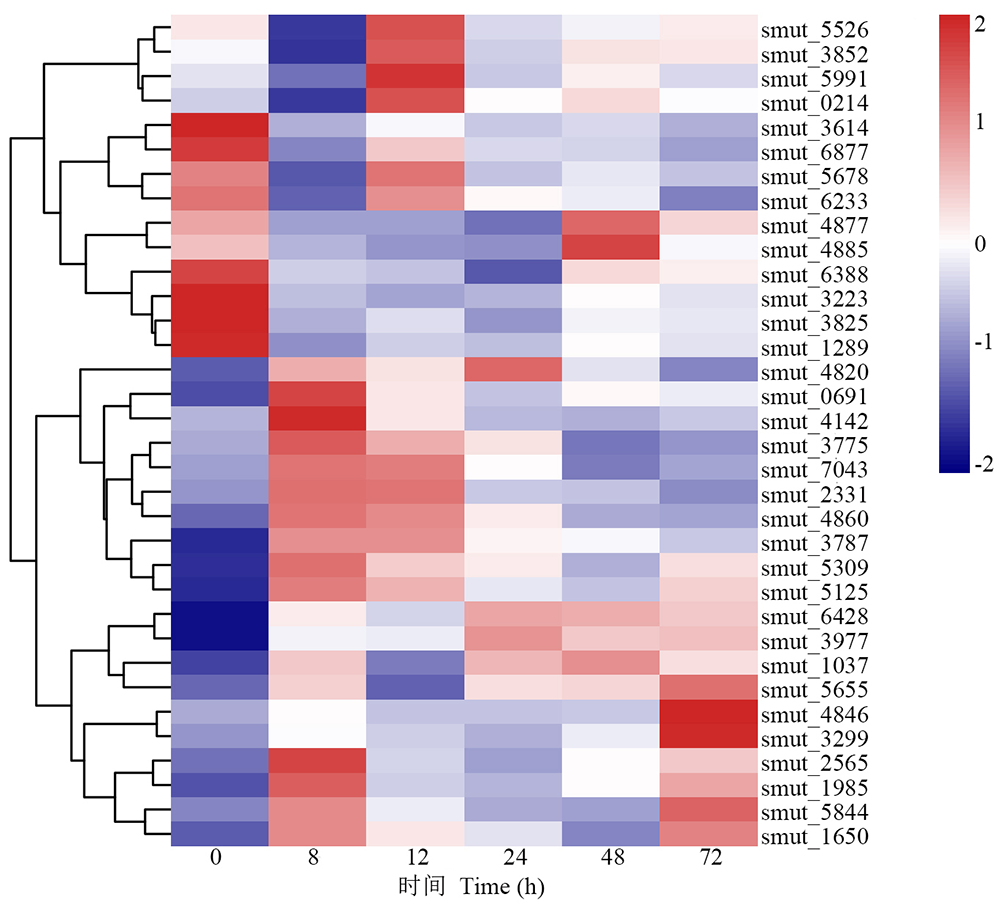 | 图6 病原相关基因在分泌蛋白中的表达模式Fig.6 Expression pattern of pathogenic related genes in secreted protein |
在稻粒黑粉病菌侵染的不同时间点, 检测到59个寄主-病原互作(pathogen-host interactions, PHIs)上调DEGs, 包括乙酰辅酶A合成酶(acetyl-CoA synthetase-like protein)编码基因smut_0340和smut_6327、PMA1-H+转运P型ATPase(PMA1-H+-transporting P-type ATPase)编码基因smut_0538、自噬相关蛋白(autophagy-related protein 12)编码基因smut_1077、转录因子ACEII(transcription factor ACEII)编码基因smut_1130、O-甲基转移酶(O-methyltransferase)编码基因smut_1293、糖苷水解酶家族蛋白(glycoside hydrolase family 18 protein)编码基因smut_1649, WD40重复蛋白(WD40 repeat-like protein)编码基因smut_3965, 几丁质酶(chitinase)编码基因smut_3971, GMC氧化还原酶(GMC oxidoreductase)编码基因smut_3977, 丝氨酸/苏氨酸蛋白激酶编码基因smut_4358, S-腺苷-L-蛋氨酸依赖性甲基转移酶(S-adenosyl-L-methionine-dependent methyltransferase)编码基因smut_4881, 糖苷水解酶家族蛋白(glycoside hydrolase family 5 protein)编码基因smut_5678(图7)。相关报道表明, 稻瘟病菌P-ATPase基因MoCTA3在稻瘟病菌侵染水稻72 h时表达量上调, 其在稻瘟病菌致病过程中起重要作用[25]; 此外, 自噬相关蛋白、几丁质酶、糖苷水解酶家族蛋白、丝氨酸/苏氨酸蛋白激酶和氧化还原酶均参与了真菌的致病性生物学过程[5, 26, 27, 28]。
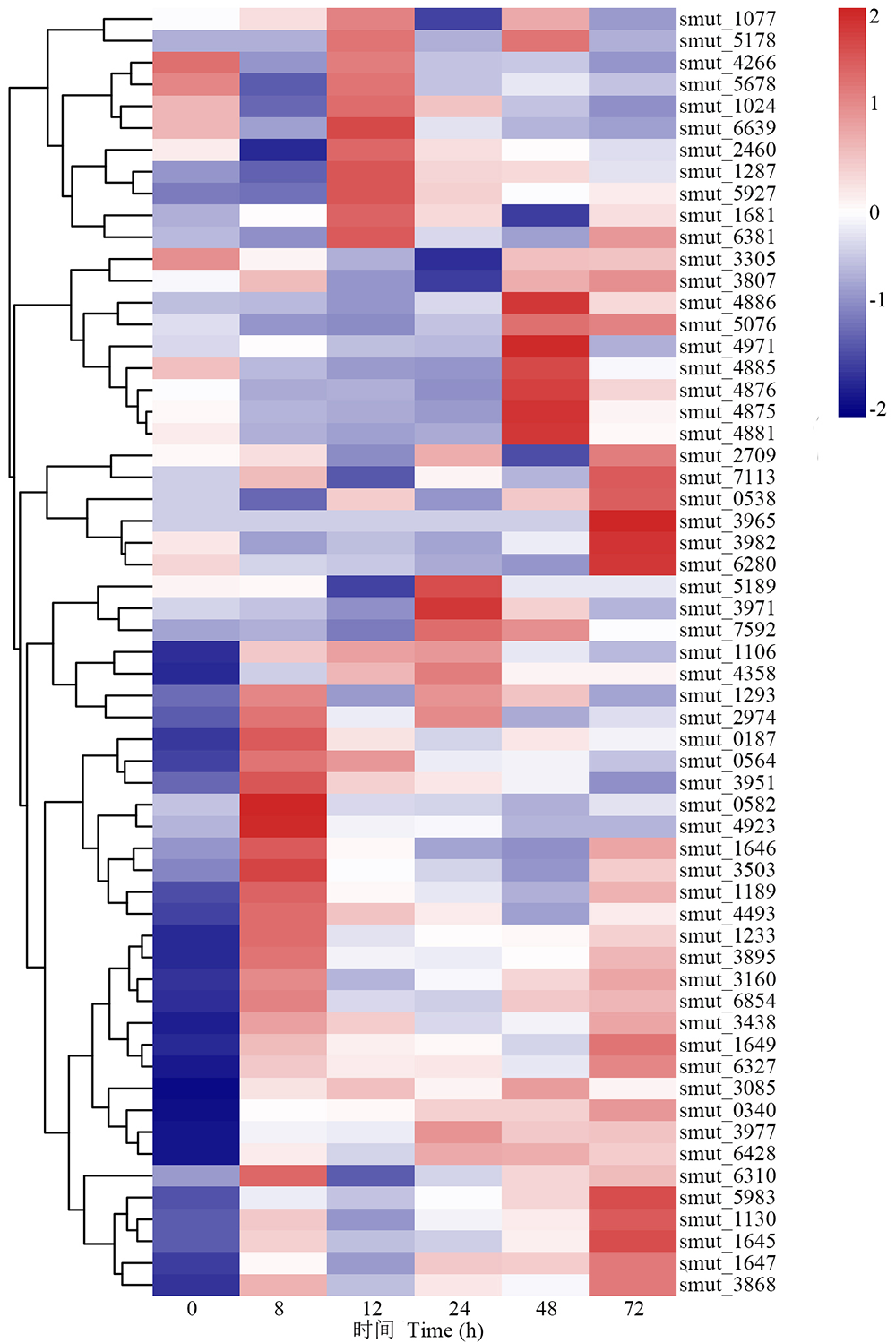 | 图7 病原相关基因在病原体-宿主相互作用中的表达模式Fig.7 Expression pattern of pathogenic related genes in pathogen-host interactions |
近年来, 稻粒黑粉病的发生对杂交稻优质、高产造成了严重的影响, 关于其防治主要依赖化学药剂, 而化学药剂的大量施用不仅会对环境造成严重污染, 且对人畜安全具有很大影响。培育抗病品种是防治该病害的首选方法之一[12]。目前, 有关抗稻粒黑粉病水稻品种报道较少, 王爱军等[5]对78份水稻不育系种质资源进行抗性筛选, 发现“ 巨丰2A” 和“ 江城3A” 对稻粒黑粉病表现抗性, 可以作为稻粒黑粉病抗病育种的重要研究材料。
为初步阐明稻粒黑粉病菌的分子致病机制, 并对防治该病害奠定一定的理论基础。本研究对稻粒黑粉菌侵染8 h的DEGs进行KEGG富集分析, 结果表明, 过氧化物酶和脂肪酸降解代谢途径显著富集。相关报道表明, 过氧化物酶和脂肪酸降解代谢在植物真菌致病过程中发挥重要作用[29]。敲除黄瓜炭疽病菌(Colletotrichum lagenarium)过氧化物酶体基因PEX6后, 病原菌的过氧化物酶代谢被阻断, 致病力明显减弱, 将突变体菌株置于葡萄糖溶液中培养, 其致病力恢复[30]。Ramos-Pamplona等[31]研究表明, 缺乏过氧化物酶体基因的稻瘟病菌(Magnaporthe oryzae) Mgpex6Delta影响具侵染能力的附着孢形成, 导致不能侵入宿主, 病原菌致病性丧失。因此, 推测过氧化物酶和脂肪酸降解代谢途径在稻粒黑粉病菌的致病信号途径中发挥重要作用。
CAZymes和分泌蛋白编码基因是真菌侵染过程中关键的致病因子。Zheng等[32]研究表明, 水稻纹枯病菌(Rhizoctonia solani)侵染宿主期间, 大量CAZymes及分泌蛋白编码基因被诱导表达, 且证实了3个分泌蛋白编码基因可引起宿主细胞的过敏性坏死反应。刘永锋等[33]鉴定了8个分泌蛋白编码基因在稻瘟病菌侵染过程中发挥关键作用。本研究对CAZymes、分泌蛋白和PHIs的DEGs进行了解析, 其中一些基因在稻粒黑粉菌侵染期间被诱导上调表达, 可能参与了稻粒黑粉病菌致病过程。如CAZymes相关基因smut_0187、smut_3124、smut_4884和smut_4358以及分泌蛋白编码基因smut_4820分别在稻粒黑粉病菌侵染8、24、48 h诱导上调表达, 证实这些基因可能是稻粒黑粉病成功侵染宿主的关键因子。
此外, 稻粒黑粉病菌侵染过程中, 自噬相关蛋白编码基因smut_1077和WD40重复蛋白编码基因smut_3965也被诱导上调表达。研究表明, 自噬蛋白介导真菌重要的侵染结构附着孢的生长[30]。灰葡萄孢(Botrytis cinereal)中自噬基因BcATG1对其发育和致病力均具有重要作用[34]。据报道许多真菌利用碳分解物阻遏(carbon catabolite repression, CCR)获取潜在碳源[35], Matar等[36]研究发现, 稻瘟病菌WD40重复蛋白编码基因MoCreC是碳分解物阻遏(carbon catabolite repression, CCR)的主要调节因子。本研究中自噬相关蛋白编码基因smut_1077和WD40重复蛋白编码基因smut_3965可能具有类似的功能, 还需进一步研究。
本研究通过转录组测序分析, 解析了稻粒黑粉病菌侵染不同时间点的基因表达差异。初步阐明过氧化物酶体和脂肪酸降解是稻粒黑粉病菌侵染宿主的关键代谢途径。并对CAZymes、分泌蛋白和PHI相关DEGs进行了解析, 其中一些DEGs可能参与了稻粒黑粉病菌的致病过程。该研究结果不仅为解析稻粒黑粉病菌的致病分子机制, 进一步防控该病害奠定了理论基础, 同时为真菌-宿主互作研究提供了新的生物学知识。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|