
{kind=link}
{kind=link}
{kind=link}
多粘类芽孢杆菌β-葡萄糖苷酶 bglA、 bglB和 bgl基因在大肠杆菌中的表达
[王艳 , 马亚茹, 万学瑞, 王川
, 马亚茹, 万学瑞, 王川* , 吴润* , 刘桂林, 刘原子, 吴自祥]
, 马亚茹, 万学瑞, 王川, 吴润, 刘桂林, 刘原子, 吴自祥]
|
|


作者简介:王艳(1991-),女,甘肃嘉峪关人,在读硕士。E-mail:409193838@qq.com
为了有效地提高β-葡萄糖苷酶的活性,本实验将多粘类芽孢杆菌( Bacillus polymyxa)β-葡萄糖苷酶 bglA、 bglB和 bgl( bglA和 bglB基因)基因分别连接在pET-28a上并在大肠杆菌C41中表达。将这3株重组菌分别命名为EA、EB及共表达菌株EAB,并将构建好的工程菌EA和EB按1∶1,1∶2,2∶1进行混合培养,分别比较单一酶组分、共表达及混合表达菌株的酶活。SDS-PAGE电泳图显示BglA和BglB的大小都为50 ku,EA和EB混合培养的蛋白大小也为50 ku,Bgl大小为100 ku,表明BglA和BglB在体外不能形成蛋白复合物,只有在生物体内才能形成Bgl复合蛋白。酶活测定结果表明共表达Bgl与多粘类芽孢杆菌的酶活差异不显著,但显著高于其他组分酶活( P<0.05)。刚果红染色结果也表明Bgl酶活比单一酶组分的酶活明显提高。本实验为纤维素酶多组分人工组装及集成生物工艺菌种的构建奠定实验基础。
To improve the enzyme activity of β-glucosidase, two β-glucosidase genes from Bacillus polymyxa were introduced separately ( bglA and bglB) and together ( bgl) into the pET-28a vector and expressed in Escherichia coli C41. The three recombinant strains were designated as EA, EB, and co-expression EAB. Strains EA and EB were mixed at ratios of 1∶1, 1∶2, and 2∶1 and their total β-glucosidase activity was compared with those of each single enzyme group, the co-expression strain, and mixed expression strains. The results of SDS-PAGE analyses showed that both BglA and BglB were 50 ku, and the size of these proteins in the EA and EB mixed cultures was also 50 ku. The size of Bgl in the co-expression strain EAB was 100 ku. These results indicated that a Bgl complex was able to form in cells, but not in vitro. In an enzyme activity assay, the activity of Bgl from the co-expression strain was not significantly different from that of bgl in B. polymyxa, but it was significantly higher than those of BglA in EA and BglB in EB ( P<0.05). The results of Congo red staining also showed that the enzyme activity of Bgl was significantly higher than those of BglA and BglB. This study lays the foundation for the construction of artificial assemblies, and for the integration of biological technologies in cellulose processing.
纤维素是植物材料的主要成分, 植物通过光合作用的形式使光能以生物能的形式固定下来, 其生成量每年高达2000亿t, 其中50%以上为纤维素和半纤维素, 这些能量相当于全球人类每年能源消耗量的20倍, 食物中所含能量的200倍, 是永远不会枯竭的可再生资源[1]。但由于纤维素具有水不溶性的高结晶构造, 其外围又被木质素层包围, 要把它水解成可利用的葡萄糖相当困难, 所以到目前为止纤维素仍没有得到很好地应用[2]。纤维素的降解包括物理法、化学法和生物降解法, 其中生物降解法因具有条件温和、成本低廉而且对环境无污染的特点备受关注。
β -葡萄糖苷酶, 又称β -D-葡萄糖苷水解酶, 属于糖苷水解酶家族3, 能将各种寡糖及纤维二糖降解为葡萄糖, 是纤维素水解糖化过程中的限速步骤。它不仅能够水解纤维二糖产生两分子的葡萄糖, 更可解除纤维二糖对内切葡聚糖酶和外切葡聚糖酶的抑制, 提高水解速率和程度。但在纤维素酶系中β -葡萄糖苷酶所占比例不足1%, 这使其成为纤维素降解成单糖的瓶颈[3]。有报道指出, 当增加纤维素酶中β -葡萄糖苷酶活性, 能有效提高纤维素的酶解效率[4]。由于不同来源的β -葡萄糖苷酶活性差异很大, 因此获得比活高、产量大、热稳定性好的β -葡萄糖苷酶一直是研究纤维素酶水解的热点问题。
有研究指出, 多粘类芽孢杆菌(Bacillus polymyxa)所产β -葡萄糖苷酶对木质纤维素来源的纤维二糖水解专一性较高, 这对于研究纤维素的酶解机制有重要意义, 因而受到国内外学者的高度重视[5]。bglA和bglB基因是β -葡萄糖苷酶bgl的两个亚基, 单个亚基不足以充分发挥水解纤维素的作用或者对纤维素的水解作用相对较弱, 因此本研究选择来源于西北特殊地理环境的多粘类芽孢杆菌作为研究材料, 并分别克隆表达了β -葡萄糖苷酶bglA及bglB基因后又将bglA和bglB基因进行了共表达。
本研究将β -葡萄糖苷酶(bgl)的两个亚基bglA和bglB分别连接到pET-28a上, 与此同时将bglA和bglB连接在pET-28a上, 将3个重组质粒分别在大肠杆菌C41中表达, 并比较重组菌株的酶活。本研究对提高β -葡萄糖苷酶的酶活及纤维素的降解具有极大的现实意义。
1.1.1 菌株与质粒
大肠杆菌DH5α 、C41及质粒pBluescript Ⅱ KS(+)、pET-28a为甘肃农业大学微生物实验室保藏, 多粘类芽孢杆菌为本实验室分离所得。
1.1.2 试剂
限制性内切酶BamHⅠ 、XhoⅠ 和SmaⅠ 、T4 DNA Ligase、T4磷酸化酶(PNK)购于TaKaRa公司; 镍柱购于GE公司; IPTG、X-gal、氨苄青霉素、卡那霉素、FastPfu fly DNA Polymerase购于北京全式金生物技术有限公司; 刚果红、水杨苷、CMC-Na购于国产分析纯; CTAB购于OXOID公司产品; 质粒DNA小提试剂盒、胶回收试剂盒购于天根生化科技有限公司。
1.1.3 培养基
培养大肠杆菌DH5α 、C41及多粘类芽孢杆菌的培养基为LB(Luria-Bertani)培养基[3], LB-微晶纤维素钠培养基用于多粘类芽孢杆菌的诱导, 2× YT(2× Yeast Tryptone)培养基用于重组菌株的诱导, LB-CMC培养基[4]用于刚果红试验检测β -葡萄糖苷酶的水解能力。
1.2.1 基因组DNA的提取
试验材料为多粘类芽孢杆菌, 于2015年3月14日在LB液体培养基中37 ℃进行振荡培养过夜, 收集菌体后采用CTAB法提取基因组DNA[6]。
1.2.2 重组质粒pET-28a::bglA和pET-28a::bglB的构建
重组质粒pET-28a::bglA和pET-28a::bglB的构建策略见图1A和图1B。以多粘类芽孢杆菌基因组DNA为模板, bglA-F/R为引物(表1)进行PCR 扩增bglA片段, PCR反应条件为:95 ℃预变性5 min; 95 ℃ 30 s; 64 ℃ 30 s; 72 ℃ 80 s; 共30个循环; 72 ℃延伸5 min; 以多粘类芽孢杆菌基因组DNA为模板, bglB-F/R为引物(表1)进行PCR 扩增bglB片段, PCR反应条件为:95 ℃预变性5 min; 95 ℃ 30 s; 61 ℃ 30 s; 72 ℃ 80 s; 共30个循环; 72 ℃延伸5 min。用胶回收试剂盒分别回收PCR产物后, 将目的片段与pET-28a同时经BamHⅠ 和XhoⅠ 酶切, 酶切后的目的片段与pET-28a纯化后通过T4 DNA Ligase进行连接, 分别得到重组质粒pET-28a::bglA和pET-28a::bglB。重组质粒经菌液PCR和双酶切验证(BamHⅠ 和XhoⅠ )正确后, 送往金唯智生物科技有限公司测序。
| 表1 β -葡萄糖苷酶bglA和bglB基因扩增引物 Table 1 Primers of β -glucosidase gene |
1.2.3 共表达重组质粒pET-28a::bgl(pET-28a::bglA::bglB)的构建
共表达重组质粒pET-28a::bgl(pET-28a::bglA::bglB)的构建策略见图1C。首先将引物bglA-R1和bglB-F1磷酸化, 然后以多粘类芽孢杆菌基因组DNA为模板, bglA-F/R1为引物进行PCR 扩增bglA片段, PCR反应条件同上; 以多粘类芽孢杆菌基因组DNA为模板, bglB-F1/R为引物进行PCR 扩增bglB片段, PCR反应条件同上。用胶回收试剂盒分别回收bglA和bglB片段, 用BamHⅠ 酶切bglA片段, XhoⅠ 酶切bglB片段, 用BamHⅠ 和XhoⅠ 酶切pBluescript Ⅱ KS(+)后, 将酶切后的bglA、bglB与pBluescript Ⅱ KS(+)纯化后通过T4 DNA Ligase连接得到重组质粒pBluescript Ⅱ KS(+)::bgl。重组质粒通过SmaⅠ 和BamHⅠ 双酶切验证正确后, 将重组质粒pBluescript Ⅱ KS(+)::bgl与pET-28a分别用BamHⅠ 和XhoⅠ 进行酶切, 酶切产物纯化后用T4 DNA Ligase进行连接得到重组质粒pET-28a::bgl。经菌液PCR和双酶切验证(BamHⅠ 和XhoⅠ )正确后, 送往金唯智生物科技有限公司测序。
1.2.4 重组质粒的转化及培养
将测序正确的重组质粒pET-28a::bglA、pET-28a::bglB及pET-28a::bgl转化至大肠杆菌C41感受态细胞中, 在含有卡那霉素(100 μ g/mL)的LB固体培养基上37 ℃过夜培养后, 筛选阳性克隆[7]。
1.2.5 β -葡萄糖苷酶的诱导表达
将筛选出的重组菌菌落接种到3 mL含卡那霉素的LB中, 37 ℃ 220 r/min过夜培养, 按1∶ 100加入到100 mL 2× YT培养基中, 37 ℃培养2.5 h, 加入IPTG到终浓度1 mmol/L, 28 ℃低温诱导14 h。将菌液用Loading buffer洗涤两次后, 超声破碎细胞:功率292.5 W, 每超声5 s间隔5 s, 至菌液澄清后4 ℃ 8000 r/min离心30 min收集上清液, 用GE公司的Ni-NTA柱进行蛋白纯化[8]。以诱导前的重组菌株为对照进行SDS-PAGE电泳[9], 分析重组蛋白表达情况。
1.2.6 重组β -葡萄糖苷酶的定量分析和酶活力的测定
通过BCA试剂盒绘制562 nm下的蛋白浓度标准曲线, 并测定样品的OD562 nm值, 通过标准曲线, 计算样品的总蛋白浓度。
通过DNS法测定β -葡萄糖苷酶酶活[10, 11] 。酶活定义:每分钟内分解底物生成1 μ g葡萄糖所需酶量定义为1个酶活单位, 以U/mL表示。试验组取0.5 mL粗酶液(BglA为110 mg蛋白)与1 mL 1%水杨苷柠檬酸缓冲液(0.1 mol/L, pH 4.8), 于50 ℃保温60 min后, 加入1.5 mL DNS煮沸10 min, 定容至25 mL, 测定OD540 nm。同时取粗酶液煮沸灭活测OD540 nm作为对照组, 平行试验重复3次, 样品酶活值为试验组酶活值与对照组酶活值之差, 并使用SPSS(Statistical Package for the Social Sciences, version 13.0 for Windows; SPSS Inc., Chicago, IL, USA)软件进行显著性差异分析。
通过LB-CMC培养基进行刚果红染色, 并根据水解圈的大小判断β -葡萄糖苷酶的水解活性[12]。
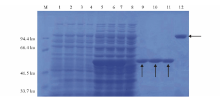
通过PCR方法克隆得到了多粘类芽孢杆菌β -葡萄糖苷酶bglA、bglB基因, 测序结果表明bglA和bglB大小分别为1361和1358 bp, 所得bglA基因序列与多粘类芽孢杆菌的bglA基因(GenBank登录号M60210.1)序列比对同源性为99%, bglB基因序列与多粘类芽孢杆菌的bglB基因(GenBank 登录号M60211.1)序列比对同源性为99%。将bglA、bglB分别连接pET-28a, 转化大肠杆菌DH5α , 构建的重组质粒pET-28a::bglA和pET-28a::bglB经BamHⅠ 和XhoⅠ 酶切验证无误后分别转化大肠杆菌C41, 质粒双酶切验证图谱如图1D, 该结果说明重组质粒pET-28a::bglA和pET-28a::bglB已成功转入大肠杆菌C41中。将得到的大肠杆菌重组菌株分别命名为EA(pET-28a::bglA)和EB(pET-28a::bglB)。
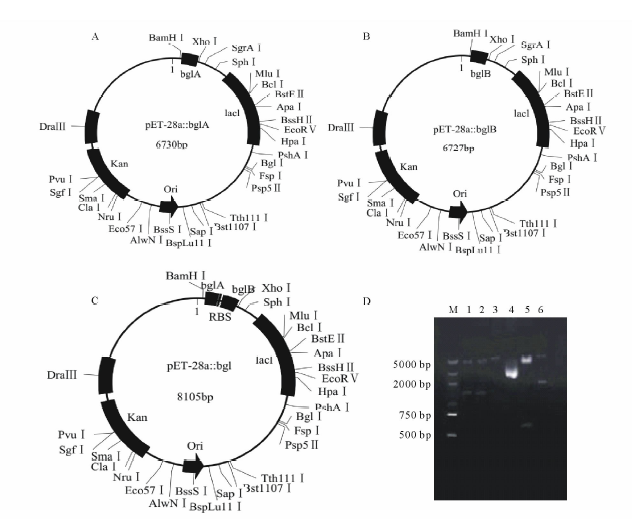 | 图1 重组质粒图谱及β -葡萄糖苷酶基因的双酶切验证 A:pET-28a::bglA重组质粒图谱Plasmid map of pET-28a::bglA; B:pET-28a::bglB重组质粒图谱Plasmid map of pET-28a::bglB; C:pET-28a::bgl重组质粒图谱Plasmid map of pET-28a:: bgl; D:β -葡萄糖苷酶基因的双酶切验证β -glucosidase gene recombinant plasmid digested; M:DNA marker; 1:pET-28a::bglA重组质粒双酶切(BamHⅠ 和XhoⅠ )pET-28a::bglA digested by BamHⅠ and XhoⅠ ; 2:pET-28a::bglB重组质粒双酶切(BamHⅠ 和XhoⅠ ) pET-28a::bglB digested by BamHⅠ and XhoⅠ ; 3:pET-28a; 4:pBluescriptⅡ KS(+); 5:pBluescriptⅡ KS(+)::bgl重组质粒双酶切(SmaⅠ 和BamHⅠ )pBluescriptⅡ KS(+)::bgl digested by SmaⅠ and BamHⅠ ; 6:pET-28a::bgl重组质粒双酶切(BamHⅠ 和XhoⅠ )pET-28a::bgl digested by BamHⅠ and XhoⅠ .Fig.1 Plasmids map and β -glucosidase gene recombinant plasmid digested from Bacillus polymyxa |
利用PCR获得bglA、bglB基因片段, 酶切连接到pBluescript Ⅱ KS(+)获得重组质粒pBluescript Ⅱ KS(+)::bgl, 通过SmaⅠ 和BamHⅠ 酶切验证无误后(图1D), 将bgl基因从pBluescript Ⅱ KS(+)::bgl上切下连接pET-28a, 获得重组质粒pET-28a::bgl, 通过BamHⅠ 和XhoⅠ 酶切验证无误后(图1D)转化大肠杆菌C41, 挑选阳性重组子提取质粒, DNA测序后证明所得片段正确, 说明共表达重组质粒pET-28a::bgl已成功转入大肠杆菌C41中。将得到的大肠杆菌共表达重组菌株命名为EAB(pET-28a::bgl)。
将EA、EB、EAB、EA与EB等体积混合的重组菌株经IPTG诱导, 超声破碎后进行纯化, 得到蛋白BglA、BglB、BglA与BglB等体积混合蛋白及Bgl, 并进行SDS-PAGE电泳检测。SDS-PAGE分析表明(图2), 诱导前无特异条带, 诱导后出现特异条带, 纯化后条带单一。诱导后的BglA、BglB、BglA与BglB等体积混合的蛋白在50 ku处有明显的蛋白表达带, 与文献中报道的多粘类芽孢杆菌所产β -葡萄糖苷酶分子量为50 ku相符[13], 诱导后的Bgl蛋白在100 ku处出现明显的蛋白表达带。根据蛋白大小可知, BglA与BglB等体积混合蛋白并没有形成完整的复合蛋白, 还是以两个相等大小的单体存在, 而诱导后的共表达蛋白Bgl在生物体内形成了一个完整的蛋白复合体。
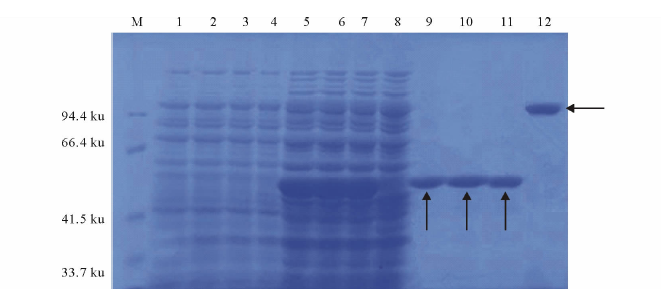 | 图2 SDS-PAGE电泳 M:蛋白Marker Protein marker; 1:EA诱导前EA before induction; 2:EB诱导前EB before induction; 3:EA和EB混合表达诱导前EA and EB cultivation by mixed proportion 1∶ 1 before induction; 4:EAB诱导前EAB before induction; 5:EA诱导后EA after induction; 6:EB诱导后EB after induction; 7:EA和EB混合表达诱导后EA and EB cultivation by mixed proportion 1∶ 1 after induction; 8:EAB诱导后EAB after induction; 9:EA纯化后 EA after purification; 10:EB纯化后 EB after purification; 11:EA和EB混合表达纯化后EA and EB cultivation by mixed proportion 1∶ 1 after purification; 12:EAB纯化后EAB after purification.EA: bglA基因连接pET-28a后转入C41感受态细胞的重组菌株Recombination strains of bglA was linked with pET-28a and expressed in Escherichia coli C41; EB: bglB基因连接pET-28a后转入C41感受态细胞的重组菌株Recombination strains of bglB was linked with pET-28a and expressed in Escherichia coli C41; EAB: bglA, bglB基因连接pET-28a后转入C41感受态细胞的重组菌株Recombination strains of bglA, bglB was linked with pET-28a and expressed in Escherichia coli C41. 下同 The same below.Fig.2 SDS-PAGE electrophoresis |
用DNS法测定重组β -葡萄糖苷酶酶活值见表2。通过SPSS软件分别比较Bgl与多粘类芽孢杆菌、BglA、BglB、EA和EB(1∶ 1)、EA和EB(1∶ 2)及EA和EB(2∶ 1)的酶活值。结果显示, Bgl与多粘类芽孢杆菌的酶活值差异不显著(P> 0.05), 但显著高于BglA、BglB、EA和EB(1∶ 1)、EA和EB(1∶ 2)及EA和EB(2∶ 1)的酶活值(P< 0.05), 是由于Bgl的bglA和bglB两个亚基形成了完整的复合蛋白, 因此酶活值大大提高; 而EA和EB混合表达的酶活值与单个表达的酶活值差异不显著, 是由于EA和EB混合培养时bglA和bglB两个亚基没有形成完整的复合蛋白。
| 表2 DNS法测定酶活值 Table 2 DNS method for the determination of enzyme activity U/mL |
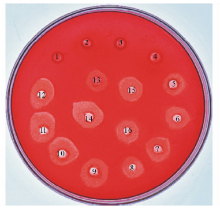
将样品按顺序加入CMC-Na的LB平板孔中, 37 ℃过夜培养, 经刚果红染色、NaCl溶液脱色后出现水解圈, 根据水解圈直径的大小即可判断水解活性的强弱。结果显示(图3), EA上清、EB上清、EAB上清、EA和EB(1∶ 1)混合表达上清没有出现水解圈, 说明在上清中不存在蛋白, 不能将蛋白分泌到细胞外。超声破碎后样品的水解圈大于纯化后蛋白样品的水解圈, 是因为破碎后蛋白含有许多杂蛋白, 而纯化后的蛋白只含有单一的蛋白条带, 因此形成的水解圈更小。将BglA和BglB按1∶ 1, 2∶ 1, 1∶ 2混合后形成的水解圈, 差别并不明显, 但将共表达蛋白Bgl加入到平板孔中, 发现所产生的水解圈大于混合表达及单一酶组分所产生的水解圈, 说明共表达β -葡萄糖苷酶Bgl的水解活性最强。
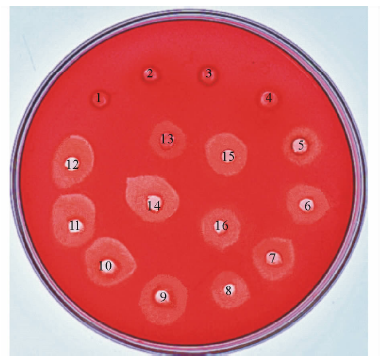 | 图3 刚果红染色水解圈 1:EA上清EA supernatant; 2:EB上清EB supernatant; 3:EA和EB(1∶ 1)混合上清EA and EB 1∶ 1 supernatant; 4:共表达上清EAB supernatant; 5:EA纯化后蛋白EA protein after purification; 6:EB纯化后蛋白EB protein after purification; 7:EA和EB(1∶ 1)混合表达纯化后蛋白EA and EB 1∶ 1 protein after purification; 8:EA破碎后EA after breaking; 9:EB破碎后EB after breaking; 10:EA和EB(1∶ 1)混合表达破碎后EA and EB 1∶ 1 after breaking; 11:共表达破碎后EAB after breaking; 12:共表达纯化后蛋白EAB protein after purification; 13:BglA和BglB(1∶ 1)BglA and BglB 1∶ 1; 14:多粘类芽孢杆菌B. polymyxa; 15:BglA和BglB(2∶ 1)BglA and BglB 2∶ 1; 16:BglA和BglB(1∶ 2)BglA and BglB 1∶ 2.Fig.3 Congo red stain hydrolysis circle |
β -葡萄糖苷酶作为纤维素降解过程中的一个重要酶组分, 在医疗、食品、生物质转化等领域具有重要的应用价值, 特别是随着近年来环境能源等危机的加重, 纤维素作为自然界最广泛的碳源受到了各国政府的高度重视。本试验采用多粘类芽孢杆菌为试验材料, 克隆表达了β -葡萄糖苷酶bglA、bglB及bgl基因, 希望能够解决β -葡萄糖苷酶酶活低的问题。
本实验先后将β -葡萄糖苷酶bglA、bglB和bgl分别连接pET-28a, 并在大肠杆菌C41中实现了表达, 实验表明BglA、BglB和Bgl 3个蛋白都为可溶性的蛋白。将重组菌株EA、EB、EA和EB(1∶ 1)混合表达后进行SDS-PAGE实验的蛋白条带差别不大, 都在50 ku(BglA蛋白大小)的位置上有蛋白条带, 可能是EA和EB(1∶ 1)混合表达在体外不能形成完整的复合蛋白。
目前, 纤维素酶活力的测定方法缺乏一个统一标准, 所用的方法与底物各有不同, 测得的酶活差异较大, 因此不具有可比性[14]。赵云等[15]从多粘类芽孢杆菌(B. polymyxa 1.794)中克隆得到β -葡萄糖苷酶基因bglA, 将其构建在pET-28a上, 转化大肠杆菌BL21, 通过β -对硝基酚葡萄糖苷(β -pNPG)为底物测定酶活, 重组β -葡萄糖苷酶的酶活为24.7 IU/mL。胡开蕾等[16]从嗜热脱氮土壤芽孢杆菌中克隆得到bglB基因, 将该基因连接到pGEX-2TL上并在大肠杆菌BL21(DE3)中表达, 通过β -pNPG为底物测定酶活, 在最适反应条件下该酶比活力为0.043 IU/mg。为了更方便的比较酶活, 本实验使用国内较便宜的水杨苷(salicin)作底物, 也能准确地反映β -葡萄糖苷酶的活力[17]。BglA与BglB单一酶组分用DNS法比较酶活时, BglA的酶活值比BglB的大, Gonzalez等[13]研究表明来自多粘类芽孢杆菌的bglA基因编码的β -葡萄糖苷酶相比bglB基因对纤维二糖底物的水解作用更强。将EA、EB诱导产生的蛋白纯化后, 其酶活值较低, 且水解圈也不明显, 究其原因可能是在IPTG诱导时蛋白合成速度较快, 空间折叠不充分而影响其活性, 也可能是与大肠杆菌C41胞内成分发生了协同作用, 研究表明错误折叠的重组蛋白与其正确折叠的蛋白相比, 其酶活显著下降[18]。而影响其酶活最主要的原因是BglA和BglB没有形成复合蛋白, 试验表明, 当BglA和BglB形成完整的复合蛋白后才能更好地发挥水解活性。目前大部分研究是将β -葡萄糖苷酶和其他的酶进行共表达, 王远等[19]将β -葡萄糖苷酶基因bglA和bglB分别与内切葡聚糖酶基因celA在枯草芽孢杆菌WB800中共表达, 共表达重组菌WB800(pP43JM2-celAbglA)的胞内酶液与纤维二糖底物作用释放出317.4 mg/L的葡萄糖, 其胞外酶液和纤维二糖底物反应释放出24 mg/L的葡萄糖; 唐自钟等[20]将β -葡萄糖苷酶基因和内切葡聚糖苷酶基因在大肠杆菌BL21(DE3)中共表达, 酶活力可达1196.8 U/mL。
本研究将β -葡萄糖苷酶两个亚基bglA和bglB在大肠杆菌C41中共表达后, 其酶活比单一酶组分及混合表达的酶活提高了4倍, 与原始菌株酶活差别不显著, 这一研究结果对纤维素降解及基因的共表达提供了实验材料。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|