

ISSN 1004-5759 CN 62-1105/S
草业学报 ›› 2023, Vol. 32 ›› Issue (7): 96-108.DOI: 10.11686/cyxb2022427
张振粉1,2( ), 黄荣1,2, 李向阳1,2, 姚博1,2, 赵桂琴1,2
), 黄荣1,2, 李向阳1,2, 姚博1,2, 赵桂琴1,2
收稿日期:2022-10-27
修回日期:2022-11-26
出版日期:2023-07-20
发布日期:2023-05-26
通讯作者:
张振粉
作者简介:张振粉(1984-),男,福建霞浦人,副教授,博士。E-mail: zhangzf@gsau.edu.cn基金资助:
Zhen-fen ZHANG1,2(), Rong HUANG1,2, Xiang-yang LI1,2, Bo YAO1,2, Gui-qin ZHAO1,2
Received:2022-10-27
Revised:2022-11-26
Online:2023-07-20
Published:2023-05-26
Contact:
Zhen-fen ZHANG
摘要:
采用高通量测序法,分析 7 份来自不同地区的燕麦种带细菌群落的细菌丰度、Alpha多样性、Beta多样性和物种组成差异,并采用 PICRUSt和FAPROTAX功能预测的方法分析了各种带细菌间的丰度差异。结果显示:1)7份燕麦中共获得5187个细菌可操作分类单元(OTUs),主要归属于33个门、180个目和435个属。2)基于OTUs的丰度及注释信息,7份燕麦种带细菌群落中变形菌门、绿弯菌门、酸杆菌门、放线菌门和蓝菌门为优势菌门,立克次氏体目和鞘脂单胞菌目为优势菌目,鞘氨醇单胞菌属、Solibacter和假节杆菌属为优势菌属。3) Alpha多样性和Beta多样性表明不同燕麦品种的细菌群落多样性和群落结构具有较大差异,其中 LXY-5 具有最大的Alpha多样性。LEFSe分析更进一步显示不同的燕麦品种生物标记的物种不同。4) PICRUSt 和 FAPROTAX 功能预测分析表明,燕麦种带细菌群落主要有代谢、有机系统和人类疾病3个代谢通路,分属于有42个 KEGG 和24个COG的二级代谢途径;FAPROTAX功能注释后共获得52个生态功能。研究结果为明确燕麦种带细菌多样性及开发新型基因资源提供了理论依据。
张振粉, 黄荣, 李向阳, 姚博, 赵桂琴. 基于Illumina MiSeq高通量测序的燕麦种带细菌多样性及功能分析[J]. 草业学报, 2023, 32(7): 96-108.
Zhen-fen ZHANG, Rong HUANG, Xiang-yang LI, Bo YAO, Gui-qin ZHAO. Seed-borne bacterial diversity of oat and functional analysis based on Illumina MiSeq high-throughput sequencing[J]. Acta Prataculturae Sinica, 2023, 32(7): 96-108.
编号 Code | 燕麦品种 Oat varieties | 产地 Production region | 地理位置 Geographical location | 储存年限 Storage year (year) |
|---|---|---|---|---|
| LXY-1 | 甜燕麦 Sweet oat | 青海西宁 Xining, Qinghai | 92°35′ E, 38°26′ N | 9 |
| LXY-2 | 燕麦473 Oat 473 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-3 | 白燕7号 Baiyan No. 7 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-4 | 贝勒2代 Baler 2 | 加拿大 Canada | 121°50′ E, 43°83′ N | 5 |
| LXY-5 | 陇燕2号 Longyan No.2 | 甘肃山丹Shandan, Gansu | 101°08′ E, 38°78′ N | 6 |
| LXY-6 | 陇燕4号 Longyan No.4 | 甘肃天祝 Tianzhu, Gansu | 102°79′ E, 37°21′ N | 5 |
| LXY-7 | 陇燕5号 Longyan No.5 | 甘肃定西 Dingxi, Gansu | 104°59′ E, 35°56′ N | 5 |
表1 供试种样的来源及相关信息
Table 1 Origin and information of tested seed samples
编号 Code | 燕麦品种 Oat varieties | 产地 Production region | 地理位置 Geographical location | 储存年限 Storage year (year) |
|---|---|---|---|---|
| LXY-1 | 甜燕麦 Sweet oat | 青海西宁 Xining, Qinghai | 92°35′ E, 38°26′ N | 9 |
| LXY-2 | 燕麦473 Oat 473 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-3 | 白燕7号 Baiyan No. 7 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-4 | 贝勒2代 Baler 2 | 加拿大 Canada | 121°50′ E, 43°83′ N | 5 |
| LXY-5 | 陇燕2号 Longyan No.2 | 甘肃山丹Shandan, Gansu | 101°08′ E, 38°78′ N | 6 |
| LXY-6 | 陇燕4号 Longyan No.4 | 甘肃天祝 Tianzhu, Gansu | 102°79′ E, 37°21′ N | 5 |
| LXY-7 | 陇燕5号 Longyan No.5 | 甘肃定西 Dingxi, Gansu | 104°59′ E, 35°56′ N | 5 |
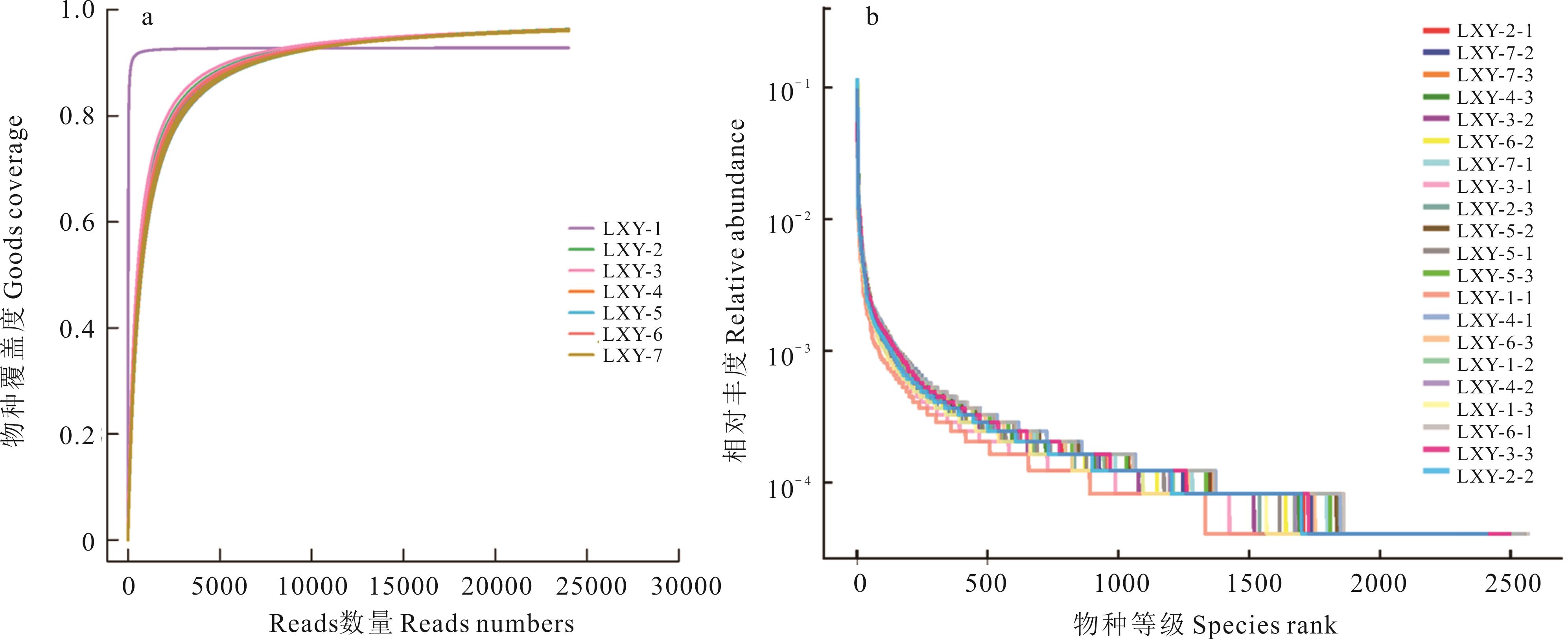
图1 燕麦种带细菌的测序深度分析
Fig.1 Deep sequencing analysis of bacteria in oat seed-borne bacteria
| 样本Sample | Chao1指数Chao1 index | Shannon指数Shannon index | Simpson指数Simpson index | 覆盖率Coverage (%) |
|---|---|---|---|---|
| LXY-1 | 2861.71±97.59b | 7.44±0.92b | 0.94±0.04a | 96.89±0.01a |
| LXY-2 | 2913.72±85.14ab | 7.98±0.43ab | 0.96±0.02a | 96.99±0.01a |
| LXY-3 | 2921.24±194.41ab | 7.83±0.74ab | 0.96±0.02a | 96.86±0.01a |
| LXY-4 | 2967.91±54.39ab | 8.56±0.45ab | 0.98±0.01a | 97.02±0.01a |
| LXY-5 | 3057.15±13.84a | 8.61±0.62ab | 0.98±0.02a | 96.94±0.01a |
| LXY-6 | 2974.10±101.12ab | 8.53±0.59ab | 0.98±0.02a | 97.00±0.01a |
| LXY-7 | 3064.78±28.90a | 8.68±0.26a | 0.98±0.01a | 96.92±0.01a |
表2 7个燕麦品种的种带细菌群落Alpha多样性指数
Table 2 Alpha diversity index of seed-borne bacterial community in 7 oat cultivars
| 样本Sample | Chao1指数Chao1 index | Shannon指数Shannon index | Simpson指数Simpson index | 覆盖率Coverage (%) |
|---|---|---|---|---|
| LXY-1 | 2861.71±97.59b | 7.44±0.92b | 0.94±0.04a | 96.89±0.01a |
| LXY-2 | 2913.72±85.14ab | 7.98±0.43ab | 0.96±0.02a | 96.99±0.01a |
| LXY-3 | 2921.24±194.41ab | 7.83±0.74ab | 0.96±0.02a | 96.86±0.01a |
| LXY-4 | 2967.91±54.39ab | 8.56±0.45ab | 0.98±0.01a | 97.02±0.01a |
| LXY-5 | 3057.15±13.84a | 8.61±0.62ab | 0.98±0.02a | 96.94±0.01a |
| LXY-6 | 2974.10±101.12ab | 8.53±0.59ab | 0.98±0.02a | 97.00±0.01a |
| LXY-7 | 3064.78±28.90a | 8.68±0.26a | 0.98±0.01a | 96.92±0.01a |
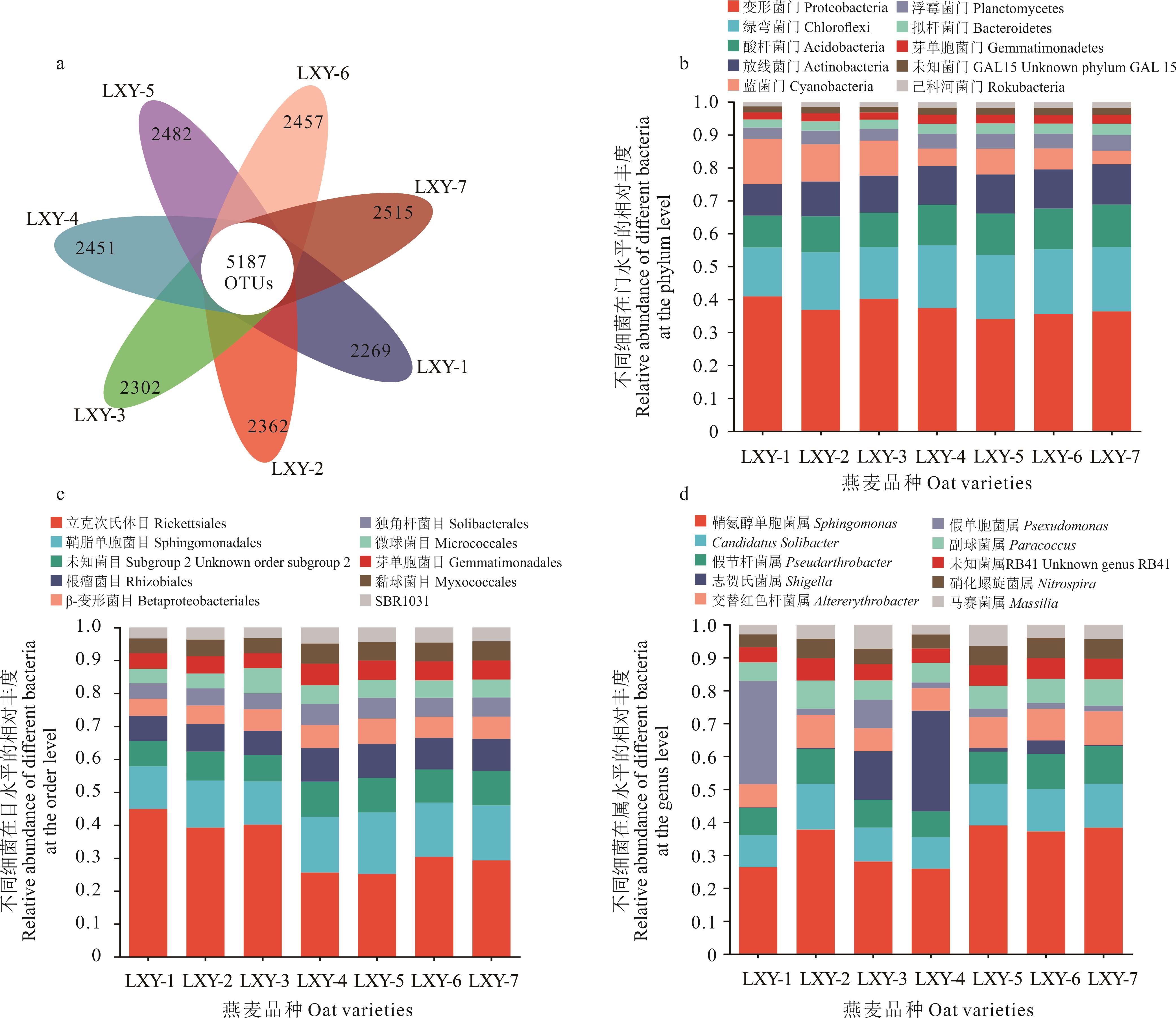
图2 燕麦种带细菌在不同分类水平的相对丰度
Fig.2 The relative abundance of oat seed-borne bacteria at different taxonomic levels
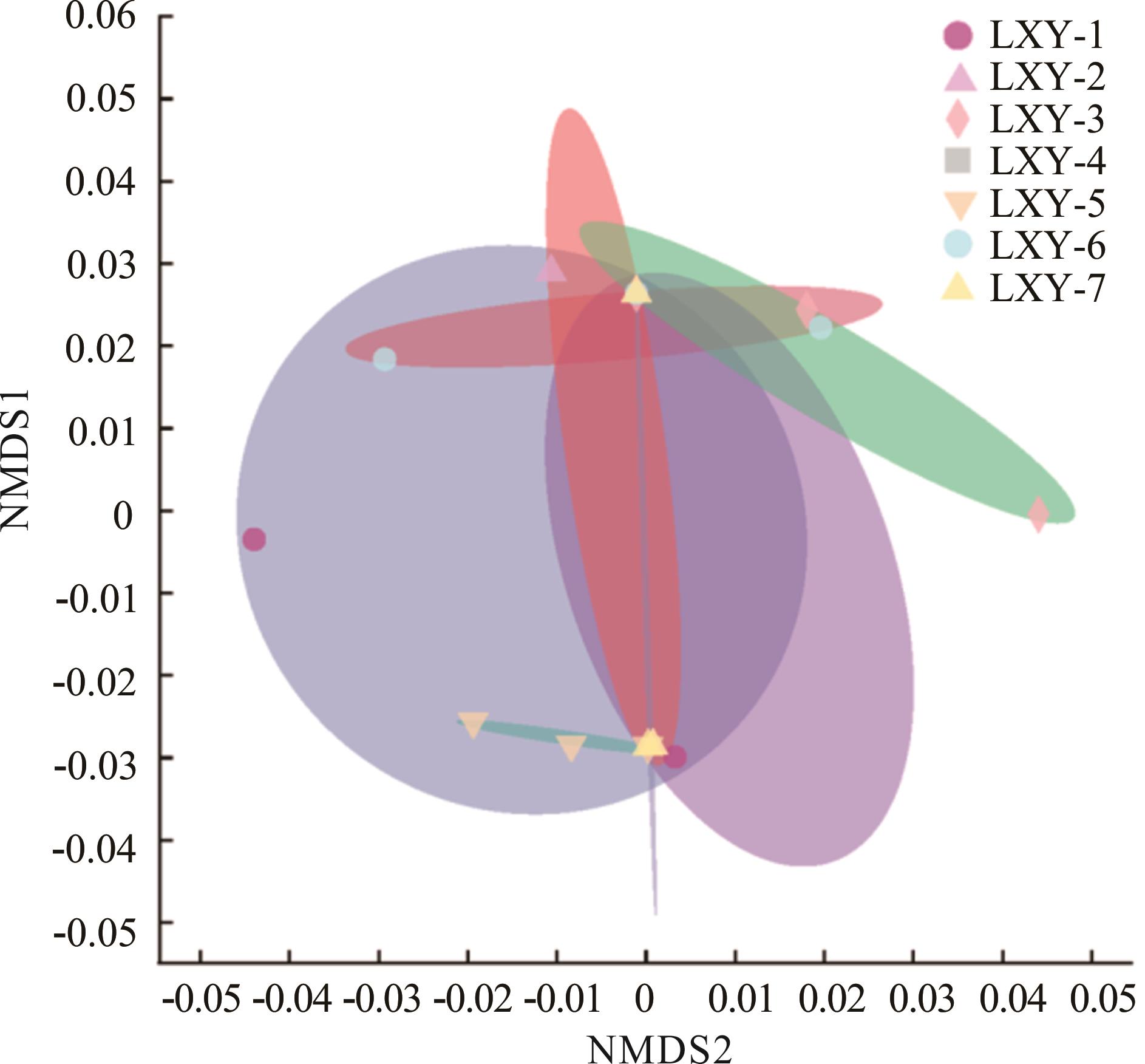
图3 7个燕麦品种的种带细菌群落结构的非度量多维尺度分析
Fig.3 NMDS of seed-borne bacterial community structure in 7 oat cultivars

图4 燕麦种带细菌线性判别分析a: 物种进化分支图Evolutionary clade of species; b: 显著差异物种 LDA 值分布柱状图,筛选标准为P<0.05, LDA值>3。P <0.05 and LDA>3 were the screening criteria for linear discriminant analysis of significantly different species.
Fig.4 Linear discriminant analysis of oat seed-borne bacteria

图5 不同燕麦品种种带细菌KEGG功能多样性热图颜色深浅表示代谢功能表达量与均值的差异程度。以同一样本基因表达量平均值为基准,高于平均值表达量则为正值,标记为红色;反之,低于平均值表达量则为负值,标记为蓝色,下同。Color shades indicate the degree of difference between the expression of metabolic function and the mean. The average value of gene expression in the same sample is used as the benchmark, and the expression above the average is positive, and the mark is red; Conversely, the expression below the average is a negative value and marked as blue, the same below.
Fig.5 Heat map of seed-borne bacteria KEGG functional diversity in different oat varieties
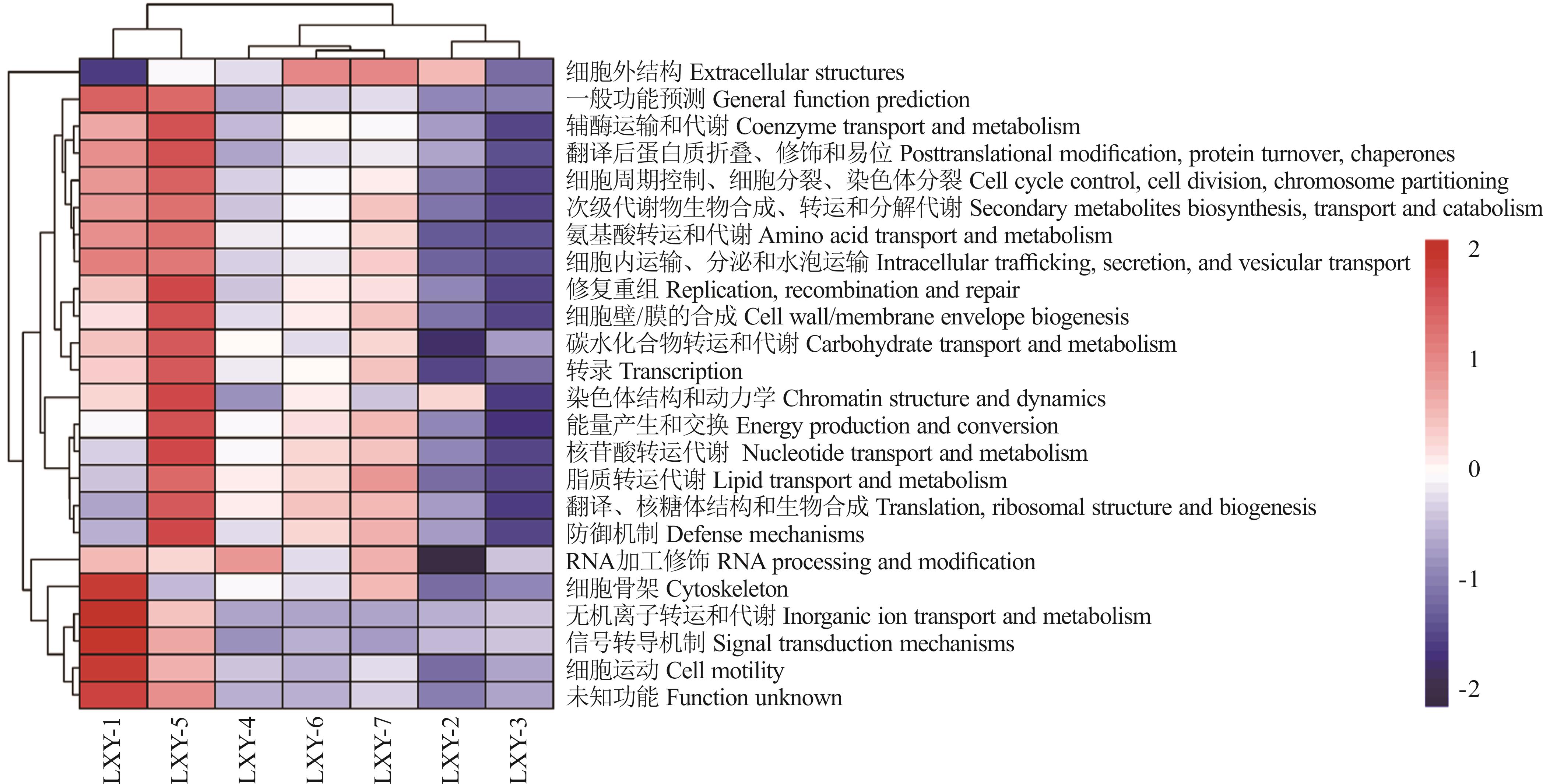
图6 不同燕麦品种种带细菌COG功能多样性热图
Fig.6 Heat map of seed-borne bacteria COG functional diversity in different oat varieties
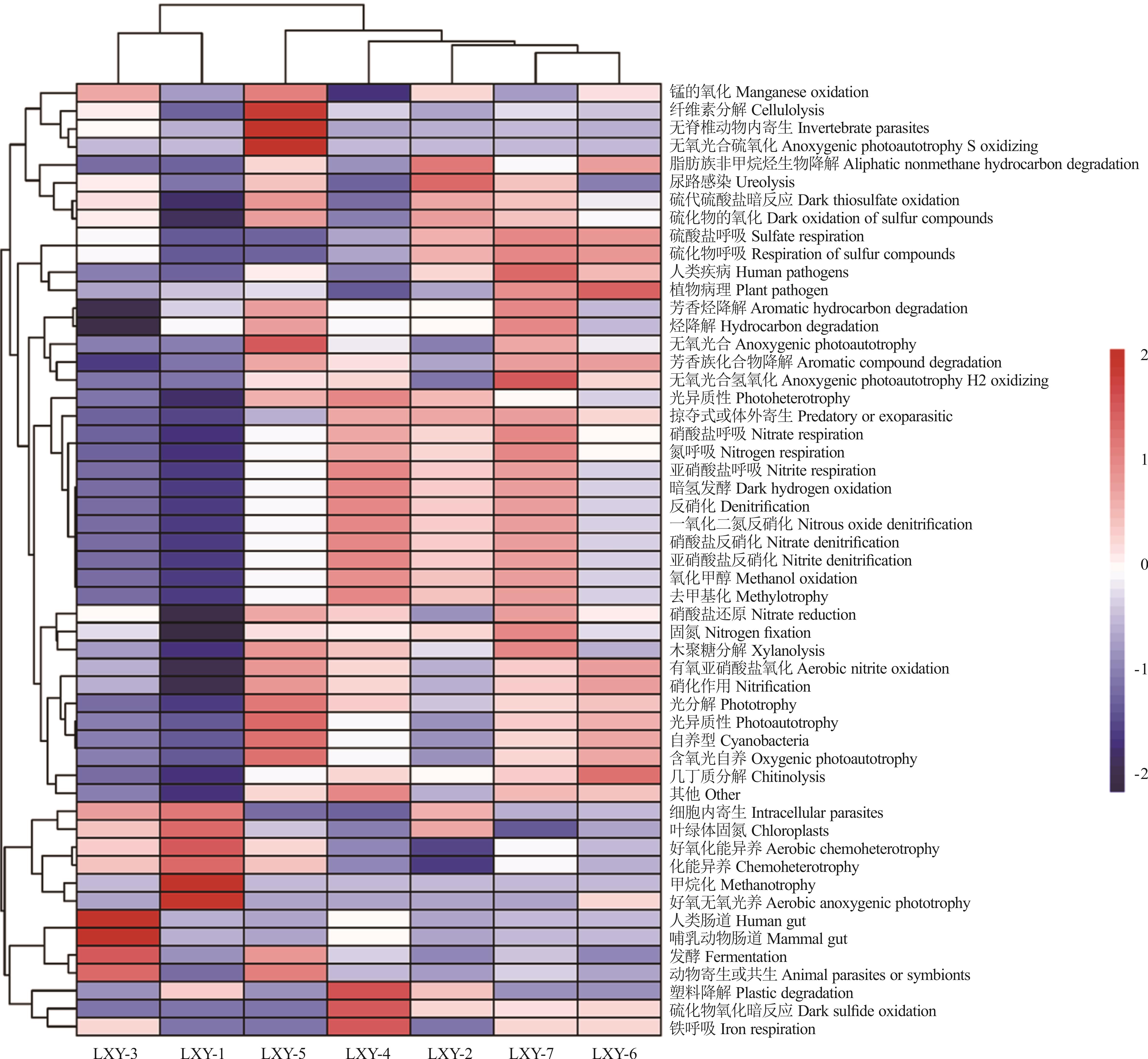
图7 不同燕麦品种种带细菌FAPROTAX功能多样性热图
Fig.7 Heat map of seed-borne bacteria FAPROTAX functional diversity in different oat varieties
| 1 | Grum M, Camloh M, Rudolph K, et al. Elimination of bean seed-borne bacteria by thermotherapy and meristem culture. Plant cell, Tissue and Organ Culture, 1998, 52(1): 79-82. |
| 2 | Nan Z B. Alfalfa disease and its comprehensive control system in China. Animal Science and Veterinary Medicine, 2001, 18(4): 81-84. |
| 南志标. 我国的苜蓿病害及其综合防治体系. 动物科学与动物医学, 2001, 18(4): 81-84. | |
| 3 | Gitaitis R, Walcott R. The epidemiology and management of seed-borne bacterial diseases. Annual Review of Phytopathology, 2007, 45(1): 371-397. |
| 4 | Trivedi P, Leach J E, Tringe S G, et al. Plant-microbiome interactions: From community assembly to plant health. Nature Reviews Microbiology, 2020, 18(11): 607-621. |
| 5 | Ahmad M, Dar Z A, Habib M. A review on oat (Avena sativa L.) as a dual-purpose crop. Scientific Research and Essays, 2014, 9(4): 52-59. |
| 6 | Boczkowska M, Tarczyk E. Genetic diversity among polish landraces of common oat (Avena sativa L.). Genetic Resource Crop and Evolution, 2013, 60(7): 2157-2169. |
| 7 | Ren C Z, Yan J T, Dong R, et al. Research progress on oat nutrients, functional properties and related products. Science and Technology of Food Industry, 2022, 43(12): 438-446. |
| 任长忠, 闫金婷, 董锐, 等. 燕麦营养成分、功能特性及其产品的研究进展. 食品工业科技, 2022, 43(12): 438-446. | |
| 8 | Ren C Z, Cui L, Yang C, et al. Establishment and application of high efficient breeding technology system of oat in China. Journal of Agricultural Science and Technology, 2016, 18(1): 1-6. |
| 任长忠, 崔林, 杨才, 等. 我国燕麦高效育种技术体系创建与应用. 中国农业科技导报, 2016, 18(1): 1-6. | |
| 9 | Hu W D, Cao X J, Wu J Y, et al. Effects of fermentation time on fermentation quality and microbial community of oat silage. Feed Research, 2022, 45(17): 106-110. |
| 胡炜东, 曹晓娟, 武俊英, 等. 发酵时间对燕麦青贮发酵品质和微生物群落的影响. 饲料研究, 2022, 45(17): 106-110. | |
| 10 | Chen K X, Li H Y. Research progress of seed endophytes and their application prospect. Chinese Wild Plant Resources, 2021, 40(11): 40-44. |
| 陈柯璇, 李海燕. 植物种子内生菌及其应用前景的研究进展. 中国野生植物资源, 2021, 40(11): 40-44. | |
| 11 | Liu L, Li T Y, Wei H, et al. Effects of a nutrient additive on the density of functional bacteria and the microbial community structure of bioorganic fertilizer. Bioresource Technology, 2014, 172: 328-334. |
| 12 | Mammoudi N, Robeson M S, Castro H F, et al. Microbial community composition and diversity in Caspian Sea sediments. FEMS Microbiology Ecology, 2015, 91(1): 1-11. |
| 13 | Xia X D, Liu X L, Wang Y, et al. Isolation and identification of spoilage organisms in whitefish. Food Science, 2015, 36(21): 175-179. |
| 夏秀东, 刘小莉, 王英, 等. 白鱼腐败细菌的分离与鉴定. 食品科学, 2015, 36(21): 175-179. | |
| 14 | Edgar R. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 2013, 10: 996-998. |
| 15 | Chen K, Chen M, Lin L, et al. Analysis of microbial community characteristics in 9 kinds of chinese herbal pieces based on 16S rRNA high throughput sequencing. Chinese Journal of Medicine Guide, 2022, 28(9): 53-56, 61. |
| 陈柯, 陈敏, 林黎, 等. 基于16S rRNA高通量测序分析9种中药饮片污染微生物群落特征. 中医药导报, 2022, 28(9): 53-56, 61. | |
| 16 | Legendre P, Legendre L. Chapter 6-Withdrawn: Multidimensional qualitative data. Developments in Environmental Modelling, 1998, 20: 207-245. |
| 17 | Wang W, Liu A, Fu W, et al. Tobacco-associated with Methylophilus sp. FP-6 enhances phytoremediation of benzophenone-3 through regulating soil microbial community, increasing photosynthetic capacity and maintaining redox homeostasis of plant. Journal of Hazardous Materials, 2022, 431: 128588. |
| 18 | Langille M G I, Zaneveld J, Caporaso J G, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 2013, 31(9): 814-821. |
| 19 | Louca S, Parfrey L W, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science, 2016, 353(6305): 1272-1277. |
| 20 | Shade A, Jacques M A, Barret M. Ecological patterns of seed microbiome diversity, transmission, and assembly. Current Opinion in Microbiology, 2017, 37: 15-22. |
| 21 | Rochefort A, Simonin M, Marais C, et al. Transmission of seed and soil microbiota to seedling. mSystems, 2021, 6(3): e00446-21. |
| 22 | Barret M, Briand M, Bonneau S, et al.Emergence shapes the structure of the seed microbiota. Applied and Environmental Microbiology, 2015, 81(4): 1257-1266. |
| 23 | Chee-Sanford J C, Li M, Davis A S, et al. Symposium do microorganisms influence seed-bank dynamics? Weed Science, 2019, 54(3): 575-587. |
| 24 | Bastias D A, Martinez-Ghersa M A, Ballaré C L, et al. Epichloë fungal endophytes and plant defenses: Not just alkaloids. Trends in Plant Science, 2017, 22(11): 939-948. |
| 25 | Truyens S, Weyens N, Cuypers A, et al. Bacterial seed endophytes: Genera, vertical transmission and interaction with plants. Environmental Microbiology, 2015, 7(1): 40-50. |
| 26 | Kim H, Lee Y H. Spatiotemporal assembly of bacterial and fungal communities of seed-seedling-adult in rice. Frontiers in Microbiology, 2021, 12: 2265. |
| 27 | Lawson C E, Wu S, Bhattacharjee A S, et al. Metabolic network analysis reveals microbial community interactions in anammox granules. Nature Communications, 2017, 8: 15416. |
| 28 | Chen Z B, Huang L, Xia Z Y, et al. Species diversity characteristics of endophytic bacteria in tobacco at different regions of Yunnan Province. Southwest China Journal of Agricultural Sciences, 2015, 28(2): 857-861. |
| 陈泽斌, 黄丽, 夏振远, 等. 云南不同地区烟草内生细菌多样性特征. 西南农业学报, 2015, 28(2): 857-861. | |
| 29 | Qessaoui R, Bouharroud R, Furze J N, et al. Applications of new rhizobacteria Pseudomonas isolates in agroecology via fundamental processes complementing plant growth. Scientific Reports, 2019, 9: 12832. |
| 30 | Matsumoto H, Fan X, Wang Y, et al. Bacterial seed endophyte shapes disease resistance in rice. Nature Plants, 2021, 7(1): 60-72. |
| 31 | Gao M L, Dong Y M, Zhang Z, et al. Effect of dibutyl phthalate on microbial function diversity and enzyme activity in wheat rhizosphere and non-rhizosphere soils. Environmental Pollution, 2020, 265 part B: 114800. |
| 32 | Zhang Z F, Shi S L. Identification and biological attributes of seed-borne bacteria isolated from lucerne cv. Gannong No.3. Acta Prataculturae Sinica, 2018, 27(1): 152-160. |
| 张振粉, 师尚礼.甘农三号紫花苜蓿种带细菌的生物功能分析及鉴定. 草业学报, 2018, 27(1): 152-160. | |
| 33 | Nannipieri P, Ascher J, Ceccherini M T, et al. Microbial diversity and soil functions. European Journal of Soil Science, 2017, 68: 12-26. |
| 34 | Guo H, Mao Z Q, Liu X L. Research progress of interaction between plant and microorganism. Chinese Agricultural Science Bulletin, 2011, 27(9): 28-33. |
| 国辉, 毛志泉, 刘训理. 植物与微生物互作的研究进展. 中国农学通报, 2011, 27(9): 28-33. | |
| 35 | Allu S, Kumar N P, Audipudi A V. Isolation, biochemical and PGP characterization of endophytic Pseudomonas aeruginosa isolated from chilli red fruit antagonistic against chilli anthracnose disease. International Journal of Current Microbiology and Applied Sciences, 2014, 3(2): 318-329. |
| 36 | Zhang R. Identification and detection of seed-born bacterium in rice. Changsha: Hunan Agricultural University, 2014. |
| 张蕊. 水稻种传细菌的检测及鉴定. 长沙: 湖南农业大学, 2014. | |
| 37 | Bastias D A, Bustos L B, Jauregui R, et al. Epichloë fungal endophytes influence seed-associated bacterial communities. Frontiers in Microbiology, 2022, 12: 795354. |
| 38 | Gao G Y. Effects of Epichloië endophyte on microbiomics in seed and seed metabolites of Achnatherum inebriant. Lanzhou: Lanzhou University, 2022. |
| 高国玉. 内生真菌对醉马草种带微生物及种子代谢物的影响. 兰州: 兰州大学, 2022. | |
| 39 | Liang Y T, Xiao X, Nuccio E E, et al. Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environmental Microbiology, 2020, 22(4): 1327-1340. |
| 40 | Xu G Q. Diversity of endophytes in Oxytropis glacialis on the Qinghai-Tibet Plateau and its adaptability to the environment. Lhasa: Tibet University, 2020. |
| 许国琪. 青藏高原冰川棘豆(Oxytropis glacialis)内生菌多样性及其与环境的适应性研究. 拉萨: 西藏大学, 2020. | |
| 41 | He X H. Dissertation analysis of endophytic microbial community structure of quinoa based on high-throughput sequencing. Chengdu: Chengdu University, 2021. |
| 何小慧. 基于高通量测序的藜麦内生微生物群落结构分析. 成都: 成都大学, 2021. | |
| 42 | Yao L A, Hu Z B, Wang L L, et al. Research development of the relationship between plant endophyte and host. Ecology and Environment, 2010, 19(7): 1750-1754. |
| 姚领爱, 胡之璧, 王莉莉, 等. 植物内生菌与宿主关系研究进展. 生态环境学报, 2010, 19(7): 1750-1754. | |
| 43 | Yan D, Zhang B T, Su A J, et al. Structural variation in the bacterial community associated with airborne particulate matter in Beijing, China, during hazy and nonhazy days. Applied and Environmental Microbiology, 2018, 84(9): e4-e18. |
| 44 | Garcfa-Garcia N, Tamames J, Linz A M, et al. Microdiversity ensures the maintenance of functional microbial communities under changing environmental conditions. The ISME Journal, 2019, 13: 2969-2983. |
| [1] | 马欣, 罗珠珠, 张耀全, 刘家鹤, 牛伊宁, 蔡立群. 黄土高原雨养区不同种植年限紫花苜蓿土壤细菌群落特征与生态功能预测[J]. 草业学报, 2021, 30(3): 54-67. |
| [2] | 史经昂, 张兵, 肖晓琳, 马晶晶, 杨向阳, 刘建秀. 结缕草肉桂醇脱氢酶基因家族全基因组序列鉴定和表达分析[J]. 草业学报, 2017, 26(6): 111-119. |
| [3] | 邹雪,张烨,吴明阳,王西瑶. 马铃薯和拟南芥GAPC酶基因的克隆及分析[J]. 草业学报, 2014, 23(1): 239-247. |
| [4] | 王丽,张俊莲,张金文,刘玉汇,白江平,余斌,杨宏羽,王蒂. 拟南芥高亲和性K+载体蛋白基因cDNA克隆及其序列特征分析[J]. 草业学报, 2013, 22(6): 230-238. |
| [5] | 李剑,张金林,王锁民,郭强. 小花碱茅HKT2;1基因全长cDNA的克隆与生物信息学分析[J]. 草业学报, 2013, 22(2): 140-149. |
| [6] | 陈婷婷,杨青川,张新全,康俊梅,丁旺,张铁军. 苜蓿乙烯应答因子基因的表达特性和生物信息学分析[J]. 草业学报, 2012, 21(6): 166-174. |
| [7] | 牛继平,张金文,王旺田,王蒂,陆艳梅,张俊莲. 马铃薯SGAs合成代谢途径末端SGT酶基因克隆及序列分析[J]. 草业学报, 2012, 21(3): 106-116. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||