

ISSN 1004-5759 CN 62-1105/S
草业学报 ›› 2026, Vol. 35 ›› Issue (4): 67-85.DOI: 10.11686/cyxb2025195
李建男1( ), 唐凯1, 孟建宇1(), 冯福应2, 赵秀娟2, 李雪菲2, 赵子怡2, 陈向阳2
), 唐凯1, 孟建宇1(), 冯福应2, 赵秀娟2, 李雪菲2, 赵子怡2, 陈向阳2
收稿日期:2025-05-19
修回日期:2025-07-01
出版日期:2026-04-20
发布日期:2026-02-07
通讯作者:
孟建宇
作者简介:Corresponding author. E-mail: meng_jianyu@imau.edu.cn基金资助:
Jian-nan LI1(), Kai TANG1, Jian-yu MENG1(), Fu-ying FENG2, Xiu-juan ZHAO2, Xue-fei LI2, Zi-yi ZHAO2, Xiang-yang CHEN2
Received:2025-05-19
Revised:2025-07-01
Online:2026-04-20
Published:2026-02-07
Contact:
Jian-yu MENG
摘要:
为解析荒漠植物根际微生物资源及其生态功能,本研究以鄂尔多斯高原沙地草原5种典型荒漠植物沙韭、杨柴、枸杞、猫头刺和大果沙棘为对象,采用三代高通量测序技术对根际土壤细菌进行全长16S rRNA基因测序,结合生物信息学分析揭示其群落特征与功能差异。结果表明:变形菌门、放线菌门和拟杆菌门为共有的优势菌门,其中属于放线菌门的类节杆菌属、节杆菌属和假节杆菌属在共现网络中具有较高连接度,构成核心微生物群落。枸杞根际呈独特的群落结构,其绿弯菌门相对丰度显著高于其他植物(P<0.05),且富集长微菌属等特异生物标志物。功能预测显示大果沙棘根际微生物碳固定通路丰度最高,而枸杞特有脂肪酸延伸通路。土壤理化分析表明有机质和速效磷是驱动枸杞根际细菌群落差异的关键因子,pH是驱动大果沙棘、猫头刺和沙韭根际细菌群落的关键因子。本研究发现荒漠植物通过特异性招募假节杆菌属等核心菌群构建抗逆功能模块,为微生物介导的荒漠生态修复提供基础研究及理论依据。
李建男, 唐凯, 孟建宇, 冯福应, 赵秀娟, 李雪菲, 赵子怡, 陈向阳. 荒漠植物根际土壤核心细菌群落及其功能研究[J]. 草业学报, 2026, 35(4): 67-85.
Jian-nan LI, Kai TANG, Jian-yu MENG, Fu-ying FENG, Xiu-juan ZHAO, Xue-fei LI, Zi-yi ZHAO, Xiang-yang CHEN. A study of the core bacterial community and its functions in the rhizosphere soil of desert plants[J]. Acta Prataculturae Sinica, 2026, 35(4): 67-85.
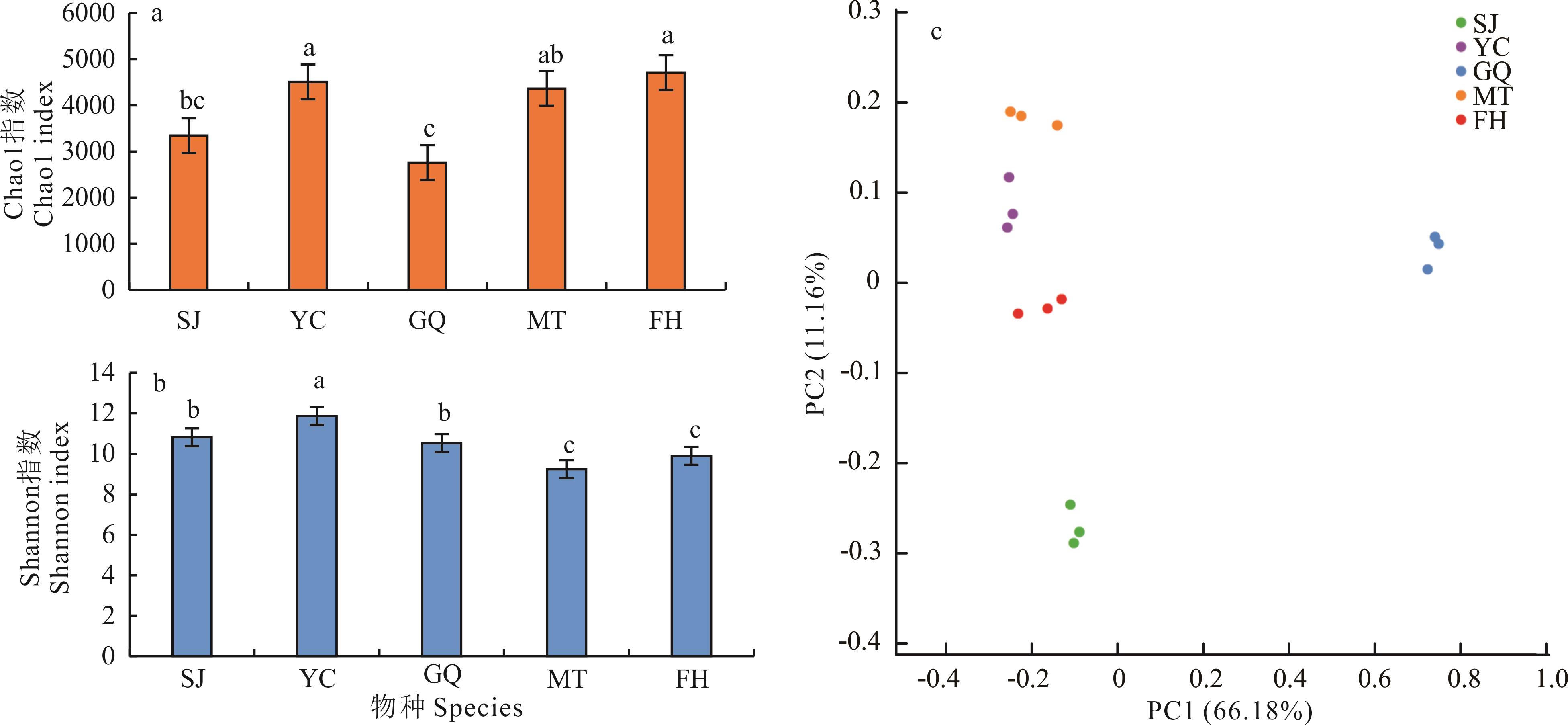
图1 5种荒漠植物根际土壤细菌群落多样性指数及PCoA分析SJ: 沙韭A. bidentatum; YC: 杨柴H. mongolicum; GQ: 枸杞L. chinense; MT: 猫头刺O. aciphylla; FH: 大果沙棘H. rhamnoides; 下同The same below; 不同小写字母表示差异显著(P<0.05) Different lowercase letters indicate significant differences (P<0.05) based on post-hoc tests; n=3.
Fig.1 Five desert plants rhizosphere soil bacterial community diversity indices and PCoA analysis

图2 5种荒漠植物根际细菌属水平群落相对丰度“其他”表示相对丰度top30以后的菌属(种) Other represents the relative abundance of bacterial genera (species) ranking below the top 30; 下同The same below.
Fig.2 Relative abundances of rhizosphere bacterial communities at the genus level for five desert plant species

图 3 5种荒漠植物根际细菌种水平群落相对丰度
Fig.3 Relative abundances of rhizospheres bacterial communities at the species level for five desert plant species

图4 5种荒漠植物根际细菌显著差异物种LDA值分布
Fig.4 The distribution of significant species LDA values of rhizosphere bacteria for five desert plant species
样品 Sample | 有机质 Organic matter (g·kg-1) | 速效磷 Available phosphorus(mg·kg-1) | 速效氮 Available nitrogen (mg·kg-1) | pH | 电导率 Electrical conductivity (μs·cm-1) |
|---|---|---|---|---|---|
| 沙韭A. bidentatum | 51.83±2.12bc | 96.92±9.62c | 4.68±1.17c | 7.93±0.15b | 295.45±7.01b |
| 杨柴H. mongolicum | 53.31±1.49b | 106.62±8.17bc | 5.62±1.12b | 7.97±0.12b | 307.62±11.38ab |
| 枸杞L. chinense | 56.82±1.81ab | 111.15±7.33b | 5.89±0.98ab | 7.84±0.15b | 328.70±9.55a |
| 猫头刺O. aciphylla | 50.38±2.35c | 98.01±9.22c | 6.11±1.23a | 8.17±0.06a | 297.32±10.41b |
| 大果沙棘H. rhamnoides | 57.92±1.24a | 121.31±5.21a | 5.12±1.29b | 7.82±0.15b | 329.88±9.28a |
表1 5种荒漠植物根际土壤理化指标
Table 1 Physicochemical indicators of rhizosphere soil for five desert plant species
样品 Sample | 有机质 Organic matter (g·kg-1) | 速效磷 Available phosphorus(mg·kg-1) | 速效氮 Available nitrogen (mg·kg-1) | pH | 电导率 Electrical conductivity (μs·cm-1) |
|---|---|---|---|---|---|
| 沙韭A. bidentatum | 51.83±2.12bc | 96.92±9.62c | 4.68±1.17c | 7.93±0.15b | 295.45±7.01b |
| 杨柴H. mongolicum | 53.31±1.49b | 106.62±8.17bc | 5.62±1.12b | 7.97±0.12b | 307.62±11.38ab |
| 枸杞L. chinense | 56.82±1.81ab | 111.15±7.33b | 5.89±0.98ab | 7.84±0.15b | 328.70±9.55a |
| 猫头刺O. aciphylla | 50.38±2.35c | 98.01±9.22c | 6.11±1.23a | 8.17±0.06a | 297.32±10.41b |
| 大果沙棘H. rhamnoides | 57.92±1.24a | 121.31±5.21a | 5.12±1.29b | 7.82±0.15b | 329.88±9.28a |
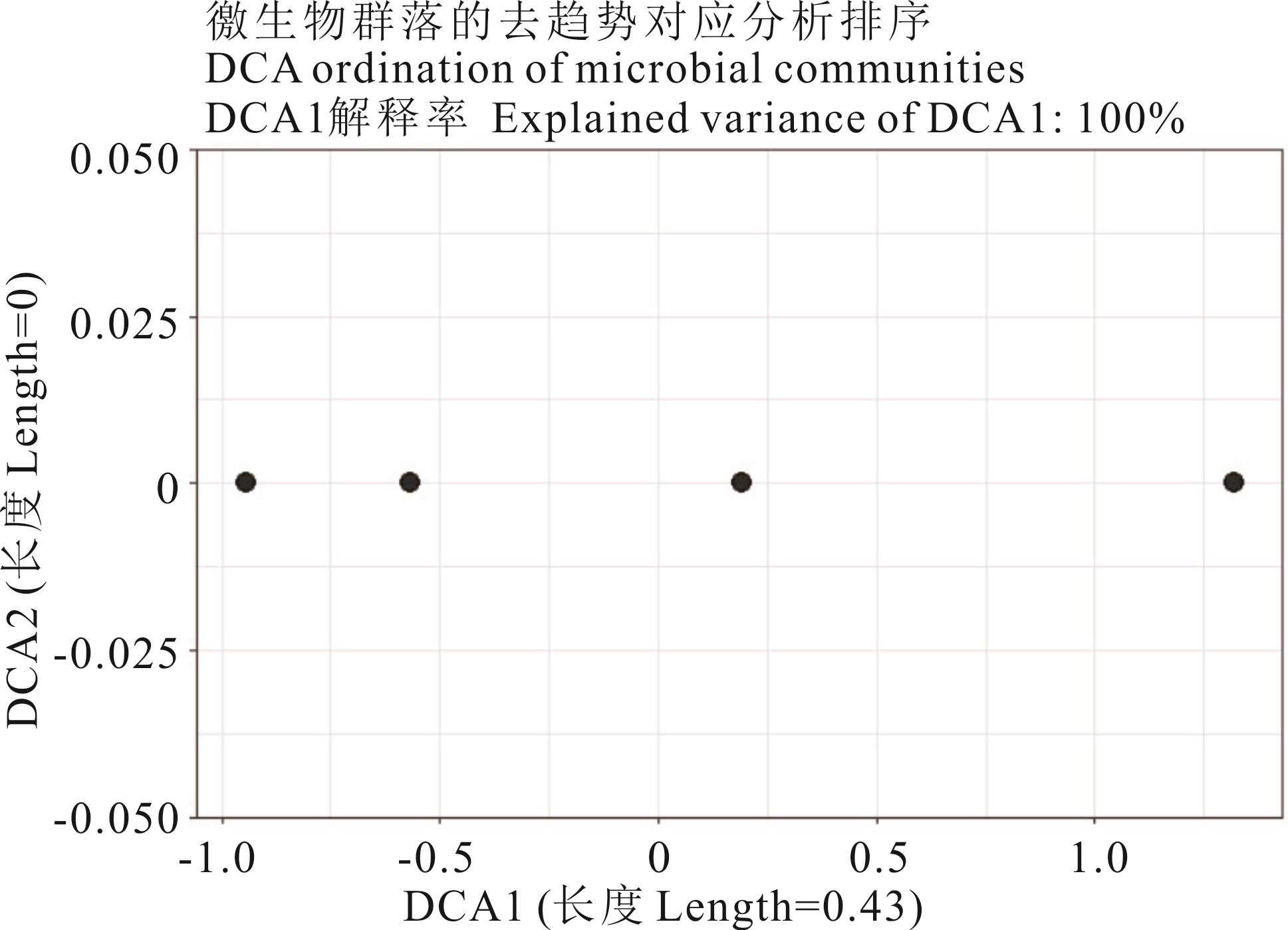
图5 5种荒漠植物根际细菌的DCA排序
Fig.5 DCA ordination of rhizosphere bacteria for five desert plant species
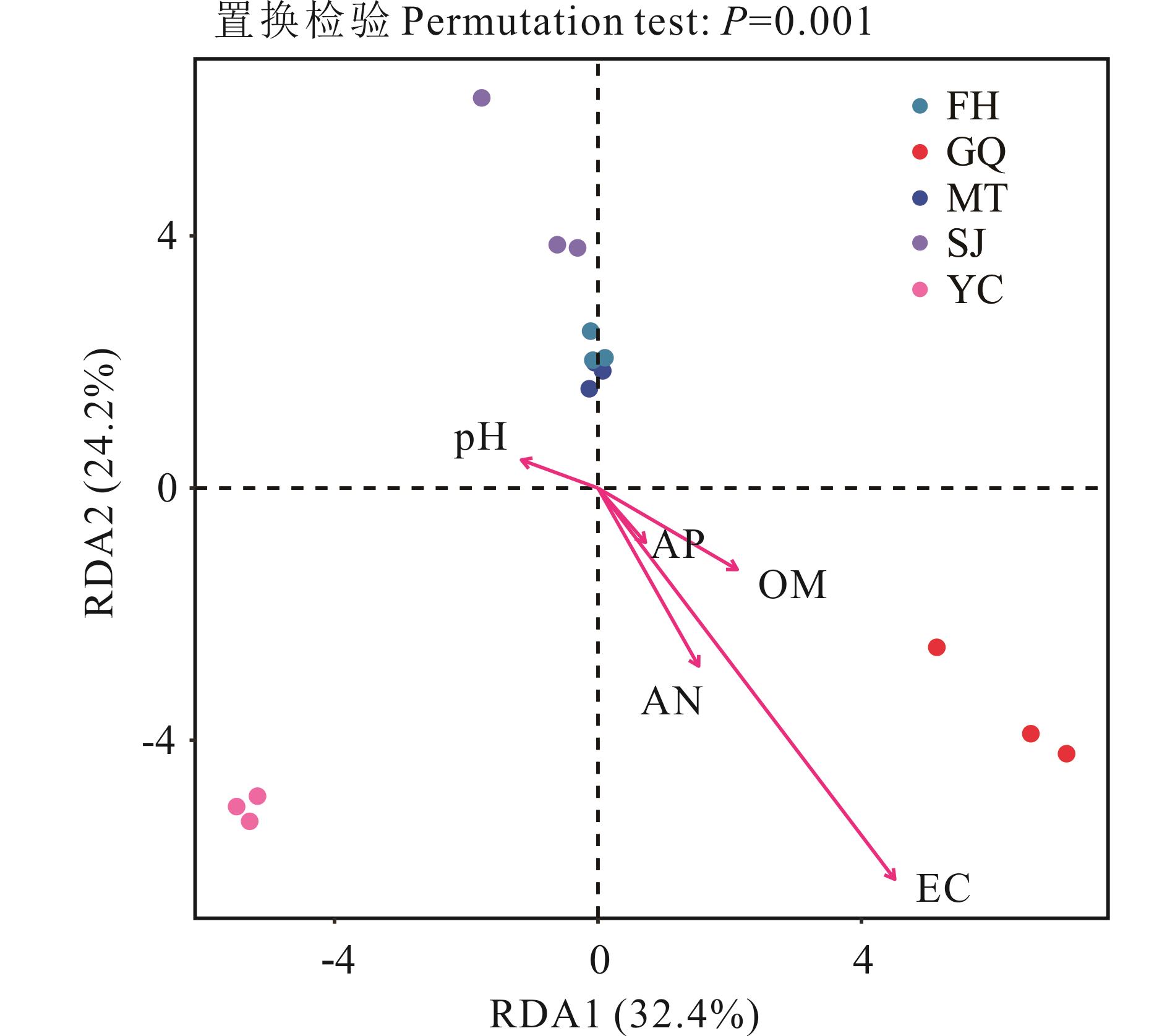
图6 5种荒漠植物根际细菌的RDA分析OM: 有机质Organic matter; AP: 速效磷Available phosphorus; AN: 速效氮Available nitrogen; EC: 电导率Electrical conductivity.
Fig.6 RDA analysis of influencing factors of rhizosphere bacterial community structure for five desert plant species

图7 5种荒漠植物根际土壤细菌核心微生物网络分析图中的节点大小代表该OTU的相对丰度高低;节点颜色越深表示该节点的degree值越大,degree则表示该节点在网络中的度,即该节点与其他节点的连接数,该值越高表明其在共现网络中的作用越大,越可能是核心群落;连线越粗表示两节点分别代表的OTU相对丰度的相关性越大,连线的颜色则表示该相关性是否显著,红色为显著,灰色为不显著。In the graph, the size of a node represents the relative abundance of the corresponding OTU; the darker the node color, the higher the degree value of that node. The degree indicates the number of connections a node has with other nodes in the network. A higher degree value suggests the node plays a more important role in the co-occurrence network and is more likely to be part of the core community; the thicker the connecting line (edge), the stronger the correlation between the relative abundances of the two OTUs represented by the nodes. The color of the line indicates whether the correlation is statistically significant: red denotes a significant correlation, while grey indicates a non-significant one.
Fig.7 Core microbial network analysis of rhizosphere bacteria for five desert plant species
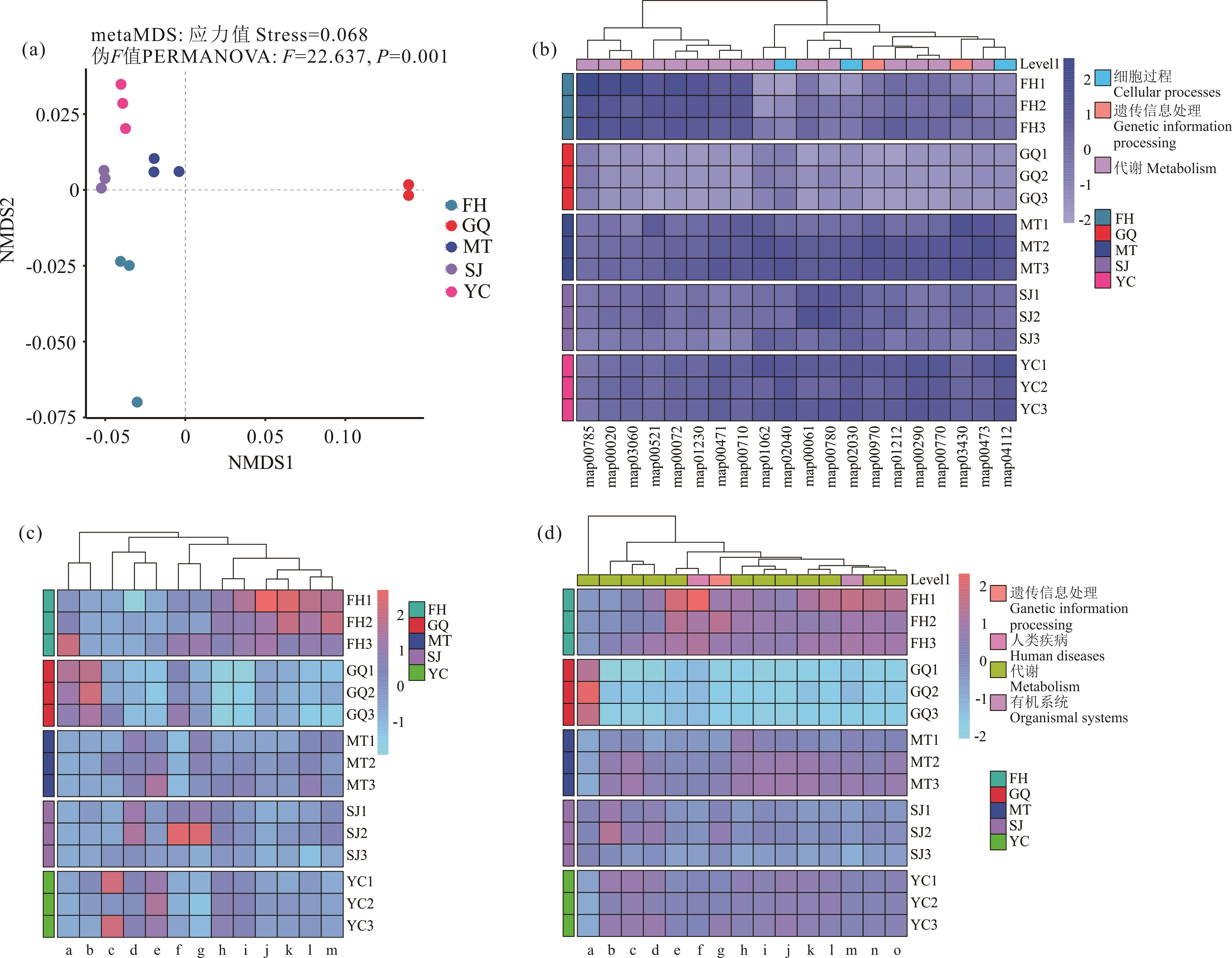
图8 5种荒漠植物根际土壤细菌功能差异分析及功能基因、酶和代谢通路热图(a): 5种荒漠植物根际土壤细菌群落功能NMDS分析Non-metric multidimensional scaling (NMDS) analysis of functional characteristics in rhizosphere soil bacterial communities of five desert plant species. (b): KEGG数据库注释的一级分类层级热图Hierarchical heatmap of primary classification levels in KEGG database annotation; map00785: 硫辛酸代谢Lipoic acid metabolism; map00020: 三羧酸循环Citrate cycle; map03060: 蛋白质分泌Protein export; map00521: 二噁英的降解Dioxin degradation; map00072: 酮体的合成与降解: Synthesis and degradation of ketone bodies; map01230: 氨基酸的生物合成Biosynthesis of amino acids; map00471: D-谷氨酰胺与D-谷氨酸代谢D-glutamine and D-glutamate metabolism; map00710: 光合生物的碳固定Carbon fixation in photosynthetic organisms; map01062: 萜类化合物与甾体化合物的生物合成Biosynthesis of terpenoids and steroids; map02040: 鞭毛组装Flagellar assembly; map00061: 脂肪酸生物合成Fatty acid biosynthesis; map00780: 生物素代谢Biotin metabolism; map02030: 细菌趋化性Bacterial chemotaxis; map00970: Apelin信号通路Apelin signaling pathway; map01212: 脂肪酸代谢Fatty acid metabolism; map00290: 亮氨酸与异亮氨酸的生物合成Leucine and isoleucine biosynthesis; map00770: 泛酸与辅酶A的生物合成Pantothenate and CoA biosynthesis; map03430: 错配修复Mismatch repair; map00473: D-丙氨酸代谢D-Alanine metabolism; map04112: 细胞循环Cell cycle. (c): KEGG数据库注释的酶丰度热图Heatmap of enzyme abundance annotated by the KEGG database; a: 蔗糖合成酶 Sucrose synthase; b: 胰蛋白酶 Trypsin; c: 过氧化物酶 Peroxidase; d: 超氧化物歧化酶 Superoxide dismutase; e: 脯氨酸羟化酶 Procollagen-proline dioxygenase; f: △1-吡咯啉-5-羧酸还原酶 Pyrroline-5-carboxylate reductase; g: 嗜热菌蛋白酶 Thermolysin; h: 过氧化氢酶 Catalase; i: 脯氨酸脱氢酶 Proline dehydrogenase; j: 蛋白酶体肽链内切酶复合体 Proteasome endopeptidase complex; k: 谷氨酰内肽酶 Glutamyl endopeptidase; l: α-淀粉酶 Alpha-amylase; m: 木糖异构酶 Xylose isomerase. (d): 根际细菌群落碳、氮、硫以及脂肪代谢功能通路热图Heatmap of carbon, nitrogen, sulfur, and lipid metabolic pathways in rhizosphere bacterial communities; a: 脂肪酸碳链延长 Fatty acid elongation; b: 脂肪酸的生物合成 Fatty acid biosynthesis; c: 脂肪酸代谢 Fatty acid metabolism; d: 不饱和脂肪酸的生物合成途径 Biosynthesis of unsaturated fatty acids; e: 叶酸介导的一碳单位池 One-carbon pool by folate; f: 肿瘤中心碳代谢 Central carbon metabolism in cancer; g: 硫转运系统 Sulfur relay system; h: 光合作用生物的碳固定 Carbon fixation in photosynthetic organisms; i: 氮素代谢 Nitrogen metabolism; j: 硫素代谢 Sulfur metabolism; k: 脂肪酸降解 Fatty acid degradation; l: 多环芳烃生物降解 Polycyclic aromatic hydrocarbon degradation; m: 近端小管碳酸氢盐重吸收 Proximal tubule bicarbonate reclamation; n: 碳素代谢 Carbon metabolism; o: 原核生物的碳固定途径 Carbon fixation pathways in prokaryotes.
Fig.8 Analysis of functional differences in rhizosphere soil bacteria of five desert plants and heatmap of functional genes, enzymes, and metabolic pathways
| [1] | Liu J H, Zhou H S, Guo Q. The effects of desertification control on the patterns of vegetation in arid and semi-arid regions of northern China. Journal of Desert Research, 2023, 43(5): 204-213. |
| 刘俊壕, 周海盛, 郭群. 中国北方干旱半干旱区沙漠化治理对植被格局的影响. 中国沙漠, 2023, 43(5): 204-213. | |
| [2] | Li Z L. Proceedings of the 2018 annual academic conference of China society of sand control and sand industry//Proceedings of the 2018 Annual Conference of the China Desertification Control and Sand Industry Association. Geermu: China Desertification Control and Sand Industry Association, 2018: 35-48. |
| 李卓玲. 内蒙古自治区荒漠化现状与防治//中国治沙暨沙业学会2018年学术年会论文集. 格尔木: 中国治沙暨沙业学会, 2018: 35-48. | |
| [3] | Wei Z J, Niu F B, Liu H M, et al. Study on response of niche of Stipa breviflora desert steppe plant populations to grazing. Chinese Journal of Grassland, 2015, 37(5): 24-32. |
| 卫智军, 牛富宝, 刘红梅, 等. 短花针茅荒漠草原植物种群生态位对放牧的响应. 中国草地学报, 2015, 37(5): 24-32. | |
| [4] | Wang C S. The analysis on forming reason and mechanism of Reaumuria soongorica community in steppe region. Hohhot: Inner Mongolia University, 2010. |
| 王常顺. 草原区红砂群落形成原因与机理分析. 呼和浩特: 内蒙古大学, 2010. | |
| [5] | Jia X H, Wu B, Yu X X, et al. Research and demonstration on key technologies for desertification control in the Beijing-Tianjin-Hebei sand source area. Acta Ecologica Sinica, 2016, 36(22): 7040-7044. |
| 贾晓红, 吴波, 余新晓, 等. 京津冀风沙源区沙化土地治理关键技术研究与示范. 生态学报, 2016, 36(22): 7040-7044. | |
| [6] | Mu W Q, Kang S M, Li P L. Advances in rhizosphere growth-promoting bacteria function on plant growth facilitation and their mechanisms. Chinese Bulletin of Life Sciences, 2022, 34(2): 118-127. |
| 穆文强, 康慎敏, 李平兰. 根际促生菌对植物的生长促进作用及机制研究进展. 生命科学, 2022, 34(2): 118-127. | |
| [7] | Li W Y, Peng Z P, Yu J H, et al. Progress and prospect on plant growth- promoting rhizobacteria of banana rhizosphere. Chinese Journal of Tropical Crops, 2011, 32(1): 182-187. |
| 李文英, 彭智平, 于俊红, 等. 香蕉根际促生菌的研究展望. 热带作物学报, 2011, 32(1): 182-187. | |
| [8] | Xu D L. Diversity of mycorrhizal and symbiotic microoganisms of six rare plants in western Ordos desert. Hohhot: Inner Mongolia University, 2021. |
| 徐道龙. 西鄂尔多斯荒漠6种珍稀植物菌根及共生微生物多样性研究. 呼和浩特: 内蒙古大学, 2021. | |
| [9] | Lucas S, Yan Z C, Martinus S, et al. Synthetic bacterial community derived from a desert rhizosphere confers salt stress resilience to tomato in the presence of a soil microbiome. The ISME Journal Emultidisciplinary Journal of Microbial Ecology, 2022, 16(8): 1907-1920. |
| [10] | Chen F B, Bai J, Lin Q Q, et al. Application of plant growth-promoting rhizobacteria(PGPR)for reducing zinc stress on paddy rice. Journal of Agro-Environment Science, 2012, 31(1): 67-74. |
| 陈佛保, 柏珺, 林庆祺, 等. 植物根际促生菌(PGPR)对缓解水稻受土壤锌胁迫的作用. 农业环境科学学报, 2012, 31(1): 67-74. | |
| [11] | Wang X Y, Bao X G, Zhang F, et al. Characteristics of bacterial community and soil enzyme activity in rhizosphere soil of desert plant Reaumuria soongorica. Acta Agrestia Sinica, 2024, 32(12): 3764-3773. |
| 王雪莹, 包新光, 张峰, 等. 荒漠植物红砂根际土壤细菌群落特征及土壤酶活性研究. 草地学报, 2024, 32(12): 3764-3773. | |
| [12] | Yang H R. Diversity and community structure of rhizospheric bacteria associated with desert shrubs in sestern Ordos. Hohhot: Inner Mongolia Agricultural University, 2016. |
| 杨鸿儒. 西鄂尔多斯荒漠灌木根际细菌多样性和群落结构的研究. 呼和浩特: 内蒙古农业大学, 2016. | |
| [13] | Zhang X X, Wu J X, Kong Z S. Cellular basis of the legume-rhizobium symbiosis. Plant Communications, 2024, 5(11):5-17. |
| [14] | Li M, Bi J T, Wang J. Bacterial community structure and key influence factors in saline soil of different sites in Ningxia. Acta Ecologica Sinica, 2020, 40(4): 1316-1330. |
| 李明, 毕江涛, 王静. 宁夏不同地区盐碱化土壤细菌群落多样性分布特征及其影响因子. 生态学报, 2020, 40(4): 1316-1330. | |
| [15] | Zhang J E. Common experimental research methods and techniques in ecology. Beijing: Chemical Industry Press, 2007. |
| 章家恩. 生态学常用实验研究方法与技术. 北京: 北京化学工业出版社, 2007. | |
| [16] | Braak C J F T, Smilauer P J. CANOCO reference manual and user’s guide to Canoco for Windows: Software for canonical community ordination (Version 4). Wageningen, Netherlands: Wageningen University & Research, 1998. |
| [17] | Sun L Y, Li G L, Zhao J, et al. Core microbiota drive multi-functionality of the soil microbiome in the Cinnamomum camphora coppice planting. BMC Microbiology, 2024, 24(1): 18. |
| [18] | Cordovez V, Dini-Andreote F, Víctor J. et al. Ecology and evolution of plant microbiomes. Annual Review of Microbiology, 2019, 73(1): 69-88. |
| [19] | Kong H G, Song G C, Ryu C M. Inheritance of seed and rhizosphere microbial communities through plant-soil feedback and soil memory. Environmental Microbiology Reports, 2019, 11(4): 479-486. |
| [20] | Patel S, Naik J H, Amaresan N. Isolation and characterization of drought resistance bacteria for plant growth promoting properties and their effect on chilli (Capsicum annuum) seedling under salt stress. Biocatalysis & Agricultural Biotechnology, 2017, 12: 85-89. |
| [21] | Etesami H, Beattie G A. Mining halophytes for plant growth-promoting halotolerant bacteria to enhance the salinity tolerance of non-halophytic crops. Front in Microbiology, 2018, 9: 1-20 |
| [22] | Nan J. Soil microbial diversity in rhizosphere of three Stipa species with substituted distribution in central and eastern steppe of Inner Mongolia. Hohhot: Inner Mongolia University, 2024. |
| 楠极. 内蒙古中东部草原替代分布的三个针茅种群的根际土壤微生物多样性. 呼和浩特: 内蒙古大学, 2024. | |
| [23] | Chen L L, Yala S X, Ru Y, et al. Characterization of soil microbes associated with a grazing-tolerant grass species, Stipa breviflora, in the Inner Mongolian desert steppe. Ecology and Evolution, 2020, 10(19): 10607-10618. |
| [24] | Fierer N, Jackson R B, Vilgalys R. The diversity and biogeography of soil bacterial communities. Proceedings of the National Academy of Sciences, 2006, 102(28): 9996-10001. |
| [25] | Zhan Y H, Yan Y L, Deng Z P, et al. The novel regulatory ncRNA, nfiS, optimizes nitrogen fixation via base pairing with the nitrogenase gene nifK mRNA in Pseudomonas stutzeri A1501. Proceedings of the National Academy of Sciences of the United States of America, 2016, 13(30): 113. |
| [26] | Peng G, Jiang Q K, Tan J, et al. Selection and field evaluation of antagonistic fungi against tobacco black shank. China Tobacco Science, 2018, 39(1): 8. |
| 彭阁, 姜乾坤, 谭军, 等. 烟草黑胫病拮抗真菌的筛选及活性评价. 中国烟草科学, 2018, 39(1): 8. | |
| [27] | Cheng L. Study on rhizosphere soil bacterial diversity of different desert steppe and major plants. Lanzhou: Lanzhou University of Technology, 2017. |
| 程琳. 不同荒漠草原和主要植物根际土壤细菌多样性研究. 兰州: 兰州理工大学, 2017. | |
| [28] | Yao J N, Dai J X,Liu S, et al. Analysis of bacterial community structure and function in rhizosphere soil of typical shrub in desert steppe of Ningxia. Acta Ecologica Sinica, 2024, 44(20): 9285-9299. |
| 姚佳妮, 代金霞, 刘爽, 等. 宁夏荒漠草原典型灌丛根际土壤细菌群落结构与功能. 生态学报, 2024, 44(20): 9285-9299. | |
| [29] | Jiang X C, Li J Y, Chen F, et al. Soil bacterial characteristics of six plant communities in the desert areas to the north of Yinshan Mountains. Arid Zone Research, 2022, 39(4): 1122-1132. |
| 蒋星驰, 李俊瑶, 陈峰, 等. 阴山北麓荒漠区6种植物群落的土壤细菌特征. 干旱区研究, 2022, 39(4): 1122-1132. | |
| [30] | Gong R H. Diversity of plant growth promoting bacteria related with primary plant dominant species in the desert steppe of Siziwang Banner. Hohhot: Inner Mongolia Agricultural University, 2024. |
| 巩瑞红. 四子王旗荒漠草原主要植物优势种的促生细菌多样性. 呼和浩特: 内蒙古农业大学, 2024. | |
| [31] | Zheng H, Qiu C, Tian H, et al. Host restriction factors against porcine epidemic diarrhea virus: A mini-review. Veterinary Research, 2025, 56(1): 1-10. |
| [32] | Meng J Y, Li H, Yang H R, et al. Diversity of phosphorus-solubilizing bacteria in rhizosphere of desert shrubs in Inner Mongolia and their phosphorus-solubilizing and siderophore-producing capabilities. Research of Environmental Sciences, 2021, 34(11): 2714-2721. |
| 孟建宇, 李蘅, 杨鸿儒, 等. 内蒙古荒漠灌木根际解磷菌多样性及其解磷和产铁载体能力. 环境科学研究, 2021, 34(11): 2714-2721. | |
| [33] | Neal A L, Shakoor A, Ruth G W, et al. Benzoxazinoids in root exudates of maize attract Pseudomonas putida to the rhizosphere. PLoS One, 2012, 7(4): 498. |
| [34] | Huo W W, Hou J Y, Gao M, et al. Correlation between abnormal leaf color phenomenon and endophytic bacteria of Loropetalum chinense var. rubrum. Guihaia, 2024, 44(10): 1827-1838. |
| 霍雯雯, 侯嘉怡, 高敏, 等. 红花檵木异常叶色现象与叶片内生细菌的相关性. 广西植物, 2024, 44(10): 1827-1838. | |
| [35] | Xie J. Screening, identification, and antagonistic mechanisms of plant pathogenic fungi antagonistic microorganisms. Chengdu: Sichuan University, 2004. |
| 谢晶. 植物病原菌拮抗微生物的筛选、鉴定及拮抗机理研究. 成都: 四川大学, 2004. | |
| [36] | Taman, Kaur J, Walia S S. Co-inoculation of indigenous Pseudomonas sp. and Priestia sp. to improve the soil health, plant growth, yield and fruit quality parameters of tomato (Solanum lycopersicum L.). Current Microbiology, 2025, 82(5): 210. |
| [37] | Lin Y, Xu Q, Xu C S, et al. Screening, identification, and fermentation condition optimization of actinobacteria antagonistic to tobacco wilt pathogen//Compilation of Excellent Papers from China Tobacco Society (2016), 2016. |
| 林勇, 徐茜, 徐辰生, 等. 烟草青枯病菌拮抗放线菌的筛选、鉴定及发酵条件优化//中国烟草学会2016年度优秀论文汇编, 2016. | |
| [38] | Yu S, He R, Song A, et al. Spatial and temporal dynamics of bacterioplankton community composition in a subtropical dammed Karst river of southwestern China. Microbiology Open, 2019, 8(2): 849. |
| [39] | Li Z, Lu L H, Guo J S, et al. Responses of spatial-temporal dynamics of bacterioplankton community to large-scale reservoir operation: A case study in the Three Gorges Reservoir, China. Scientific reports, 2017, 7: 42-46. |
| [40] | Chen G J, Zong J. Woeseia oceani Gen. nov. sp nov. a chemoheterotrophic member of the order Chromatiales, and proposal of Woeseiaceae fam. nov. International Journal of Systematic and Evolutionary Microbiology, 2016, 66: 107-112. |
| [41] | Tang M, Ren J H, Hu J J, et al. Mechanism of arbuscular mycorrhizal fungi (AMF) in enhancing the drought resistance of hippophae. Beijing: The 5th Youth Academic Annual Conference of the Chinese Forestry of Society, 2001. |
| 唐明, 任嘉红, 胡景江, 等. 丛枝菌根真菌(AMF)提高沙棘抗旱的机制. 北京: 中国林学会第五届青年学术年会, 2001. | |
| [42] | Zhao S C. Isolation, screening and promoting effect of rhizosphere soil functional strains of four endangered plants in desert. Hohhot: Inner Mongolia University, 2019. |
| 赵世超. 四种荒漠珍稀植物根际土壤功能菌株的分离筛选及其促生作用研究. 呼和浩特: 内蒙古大学, 2019. | |
| [43] | Yu Q L, Zhao Z R, Liu L. Advance in the function and regulation of rhizosphere microbiota. Journal of Microbiology, 2023, 43(5): 1-8. |
| 喻其林, 赵梓润, 刘琳. 根际微生物群落功能与调控的研究进展. 微生物学杂志, 2023, 43(5): 1-8. | |
| [44] | Yang L, Qi Y, Liu S X, et al. Structure and function of plant stress-related protein kinases. Journal of Plant Genetic Resources, 2013, 14(4): 659-667. |
| [45] | Aziz R K, Bartels D, Best A A, et al. The RAST server: Rapid annotations using subsystems technology. BMC Genomics, 2008, 9: 1-15. |
| [46] | Poole P, Ramachandran V, Terpolilli J. Rhizobia: From saprophytes to endosymbionts. Nature Reviews Microbiology, 2018, 16(5): 291-303. |
| [47] | Alori E T, Glick B R, Babalola O O. Microbial phosphorus solubilization and its potential for use in sustainable agriculture. Frontiers in Microbiology, 2017, 8: 971. |
| [48] | Barwinska-Sendra A, Garcia Y M, Sendra K M, et al. An evolutionary path to altered cofactor specificity in a metalloenzyme. Nature Communications, 2020, 11(1): 16478. |
| [49] | Fan W Q. Study on the mechanism of alfalfa recruited rhizosphere microbiome in response to drought and salt stress. Hohhot: Inner Mongolia Agricultural University, 2024. |
| 范文强. 苜蓿招募根际微生物响应干旱和盐胁迫的机制研究. 呼和浩特: 内蒙古农业大学, 2024. | |
| [50] | Wang Q W, Zhang H L, Meng Q Y, et al. Cloning and expression analysis of a Na+/H+ antiporter gene (nhaA) from Pseudomonas stutzeri. Chinese Agricultural Science Bulletin, 2010, 37(15): 19-24. |
| 王全伟, 张海玲, 孟庆英, 等. 施氏假单胞菌Na+/H+逆向转运蛋白基因nhaA的克隆与表达分析. 中国农学通报, 2010, 37(15): 19-24. | |
| [51] | Nishino S F, Spain J C. Biodegradation of 3-nitrotyrosine by Burkholderia sp. strain JS165 and Variovorax paradoxus JS171. Applied & Environmental Microbiology, 2006, 72(2): 1040-1044. |
| [1] | 邢静, 范文强, 王佳妮, 石凤翎. 干旱胁迫下 2个扁蓿豆品种根际细菌多样性及土壤灭菌对其生长的影响[J]. 草业学报, 2024, 33(12): 147-159. |
| [2] | 苗阳阳, 张艳蕊, 宋标, 刘旭桐, 张安琪, 吕金泽, 张浩, 张小华, 欧阳佳慧, 李旺, 曲善民. 碱蓬根际和内生细菌菌株对盐碱胁迫下苜蓿生长的影响[J]. 草业学报, 2022, 31(9): 107-117. |
| [3] | 马全林, 张锦春, 李得禄, 杨昊天. 腾格里沙漠植物区系特征分析[J]. 草业学报, 2020, 29(3): 16-26. |
| [4] | 李新乐, 鲍芳, 吴波, 曹艳丽, 刘明虎, 段瑞兵. 荒漠植物白刺新固定碳在植物-土壤系统中的分配[J]. 草业学报, 2019, 28(2): 33-40. |
| [5] | 白梦杰, 陶奇波, 韩云华, 王彦荣. 十种荒漠植物种子萌发对温度的响应[J]. 草业学报, 2019, 28(12): 53-62. |
| [6] | 杨景宁,王彦荣. PEG模拟干旱胁迫对四种荒漠植物种子萌发的影响[J]. 草业学报, 2012, 21(6): 23-29. |
| [7] | 杨景宁,王彦荣. NaCl胁迫对四种荒漠植物种子萌发的影响[J]. 草业学报, 2012, 21(5): 32-38. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||