
ISSN 1004-5759 CN 62-1105/S
Acta Prataculturae Sinica ›› 2023, Vol. 32 ›› Issue (7): 96-108.DOI: 10.11686/cyxb2022427
Previous Articles Next Articles
Zhen-fen ZHANG1,2( ), Rong HUANG1,2, Xiang-yang LI1,2, Bo YAO1,2, Gui-qin ZHAO1,2
), Rong HUANG1,2, Xiang-yang LI1,2, Bo YAO1,2, Gui-qin ZHAO1,2
Received:2022-10-27
Revised:2022-11-26
Online:2023-07-20
Published:2023-05-26
Contact:
Zhen-fen ZHANG
Zhen-fen ZHANG, Rong HUANG, Xiang-yang LI, Bo YAO, Gui-qin ZHAO. Seed-borne bacterial diversity of oat and functional analysis based on Illumina MiSeq high-throughput sequencing[J]. Acta Prataculturae Sinica, 2023, 32(7): 96-108.
编号 Code | 燕麦品种 Oat varieties | 产地 Production region | 地理位置 Geographical location | 储存年限 Storage year (year) |
|---|---|---|---|---|
| LXY-1 | 甜燕麦 Sweet oat | 青海西宁 Xining, Qinghai | 92°35′ E, 38°26′ N | 9 |
| LXY-2 | 燕麦473 Oat 473 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-3 | 白燕7号 Baiyan No. 7 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-4 | 贝勒2代 Baler 2 | 加拿大 Canada | 121°50′ E, 43°83′ N | 5 |
| LXY-5 | 陇燕2号 Longyan No.2 | 甘肃山丹Shandan, Gansu | 101°08′ E, 38°78′ N | 6 |
| LXY-6 | 陇燕4号 Longyan No.4 | 甘肃天祝 Tianzhu, Gansu | 102°79′ E, 37°21′ N | 5 |
| LXY-7 | 陇燕5号 Longyan No.5 | 甘肃定西 Dingxi, Gansu | 104°59′ E, 35°56′ N | 5 |
Table 1 Origin and information of tested seed samples
编号 Code | 燕麦品种 Oat varieties | 产地 Production region | 地理位置 Geographical location | 储存年限 Storage year (year) |
|---|---|---|---|---|
| LXY-1 | 甜燕麦 Sweet oat | 青海西宁 Xining, Qinghai | 92°35′ E, 38°26′ N | 9 |
| LXY-2 | 燕麦473 Oat 473 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-3 | 白燕7号 Baiyan No. 7 | 甘肃通渭 Tongwei, Gansu | 105°0l′ E, 35°23′ N | 5 |
| LXY-4 | 贝勒2代 Baler 2 | 加拿大 Canada | 121°50′ E, 43°83′ N | 5 |
| LXY-5 | 陇燕2号 Longyan No.2 | 甘肃山丹Shandan, Gansu | 101°08′ E, 38°78′ N | 6 |
| LXY-6 | 陇燕4号 Longyan No.4 | 甘肃天祝 Tianzhu, Gansu | 102°79′ E, 37°21′ N | 5 |
| LXY-7 | 陇燕5号 Longyan No.5 | 甘肃定西 Dingxi, Gansu | 104°59′ E, 35°56′ N | 5 |
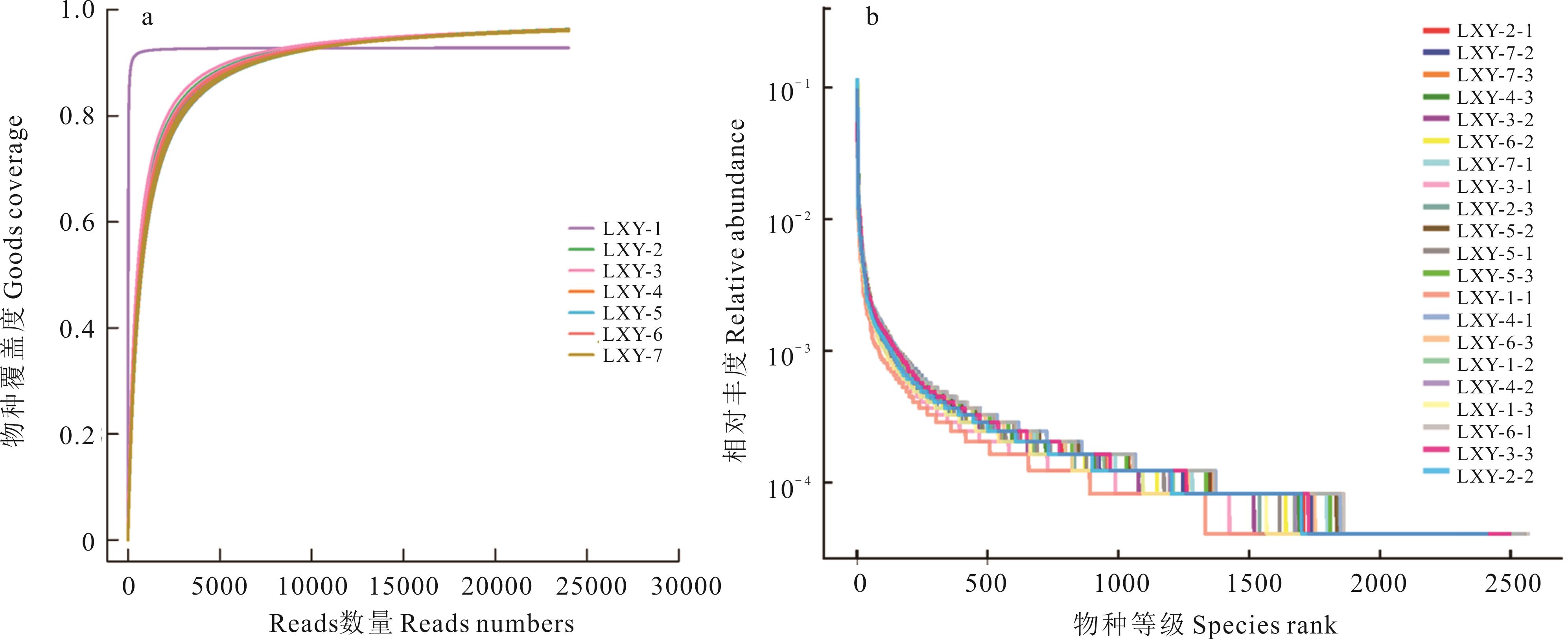
Fig.1 Deep sequencing analysis of bacteria in oat seed-borne bacteria
| 样本Sample | Chao1指数Chao1 index | Shannon指数Shannon index | Simpson指数Simpson index | 覆盖率Coverage (%) |
|---|---|---|---|---|
| LXY-1 | 2861.71±97.59b | 7.44±0.92b | 0.94±0.04a | 96.89±0.01a |
| LXY-2 | 2913.72±85.14ab | 7.98±0.43ab | 0.96±0.02a | 96.99±0.01a |
| LXY-3 | 2921.24±194.41ab | 7.83±0.74ab | 0.96±0.02a | 96.86±0.01a |
| LXY-4 | 2967.91±54.39ab | 8.56±0.45ab | 0.98±0.01a | 97.02±0.01a |
| LXY-5 | 3057.15±13.84a | 8.61±0.62ab | 0.98±0.02a | 96.94±0.01a |
| LXY-6 | 2974.10±101.12ab | 8.53±0.59ab | 0.98±0.02a | 97.00±0.01a |
| LXY-7 | 3064.78±28.90a | 8.68±0.26a | 0.98±0.01a | 96.92±0.01a |
Table 2 Alpha diversity index of seed-borne bacterial community in 7 oat cultivars
| 样本Sample | Chao1指数Chao1 index | Shannon指数Shannon index | Simpson指数Simpson index | 覆盖率Coverage (%) |
|---|---|---|---|---|
| LXY-1 | 2861.71±97.59b | 7.44±0.92b | 0.94±0.04a | 96.89±0.01a |
| LXY-2 | 2913.72±85.14ab | 7.98±0.43ab | 0.96±0.02a | 96.99±0.01a |
| LXY-3 | 2921.24±194.41ab | 7.83±0.74ab | 0.96±0.02a | 96.86±0.01a |
| LXY-4 | 2967.91±54.39ab | 8.56±0.45ab | 0.98±0.01a | 97.02±0.01a |
| LXY-5 | 3057.15±13.84a | 8.61±0.62ab | 0.98±0.02a | 96.94±0.01a |
| LXY-6 | 2974.10±101.12ab | 8.53±0.59ab | 0.98±0.02a | 97.00±0.01a |
| LXY-7 | 3064.78±28.90a | 8.68±0.26a | 0.98±0.01a | 96.92±0.01a |
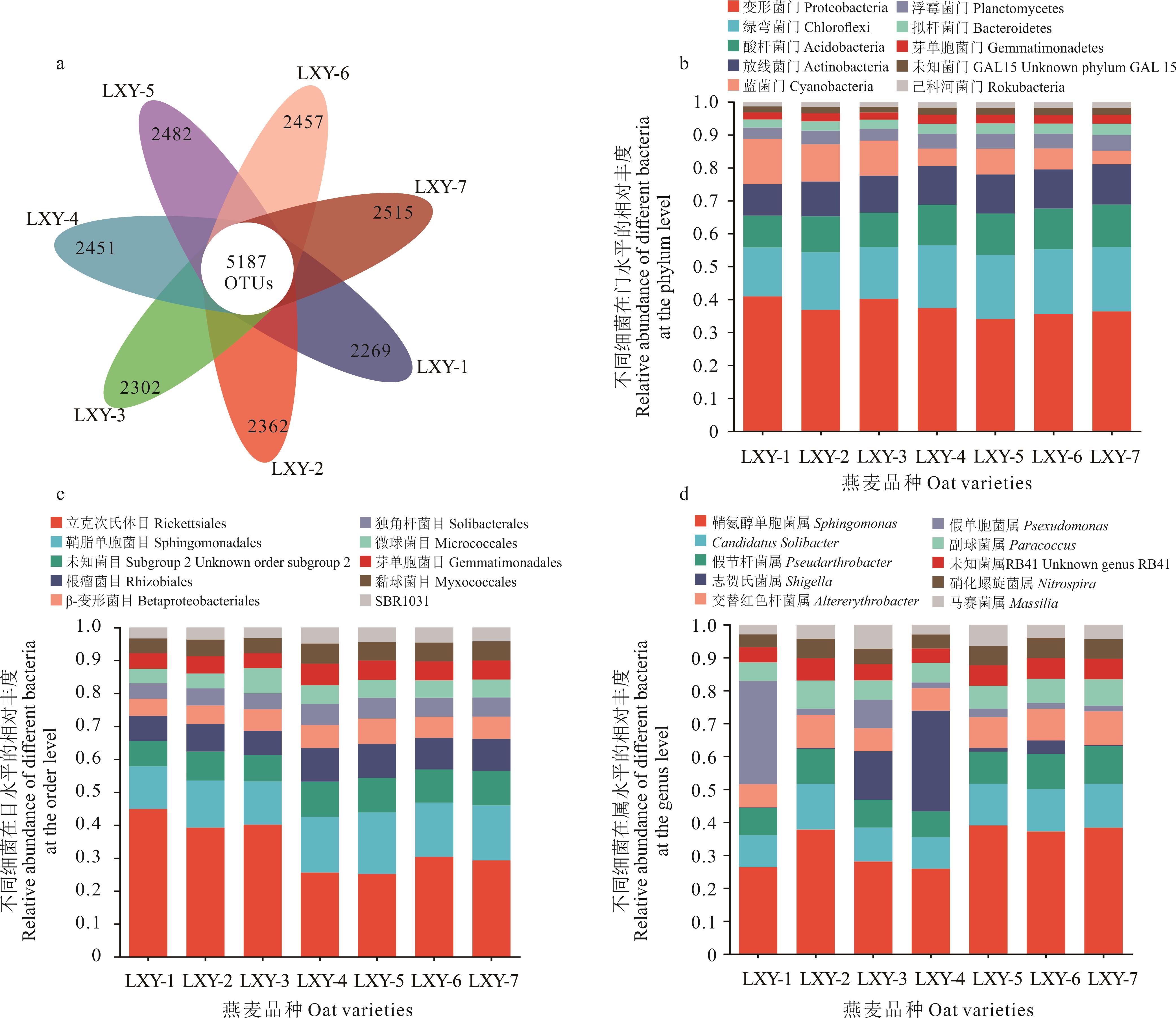
Fig.2 The relative abundance of oat seed-borne bacteria at different taxonomic levels
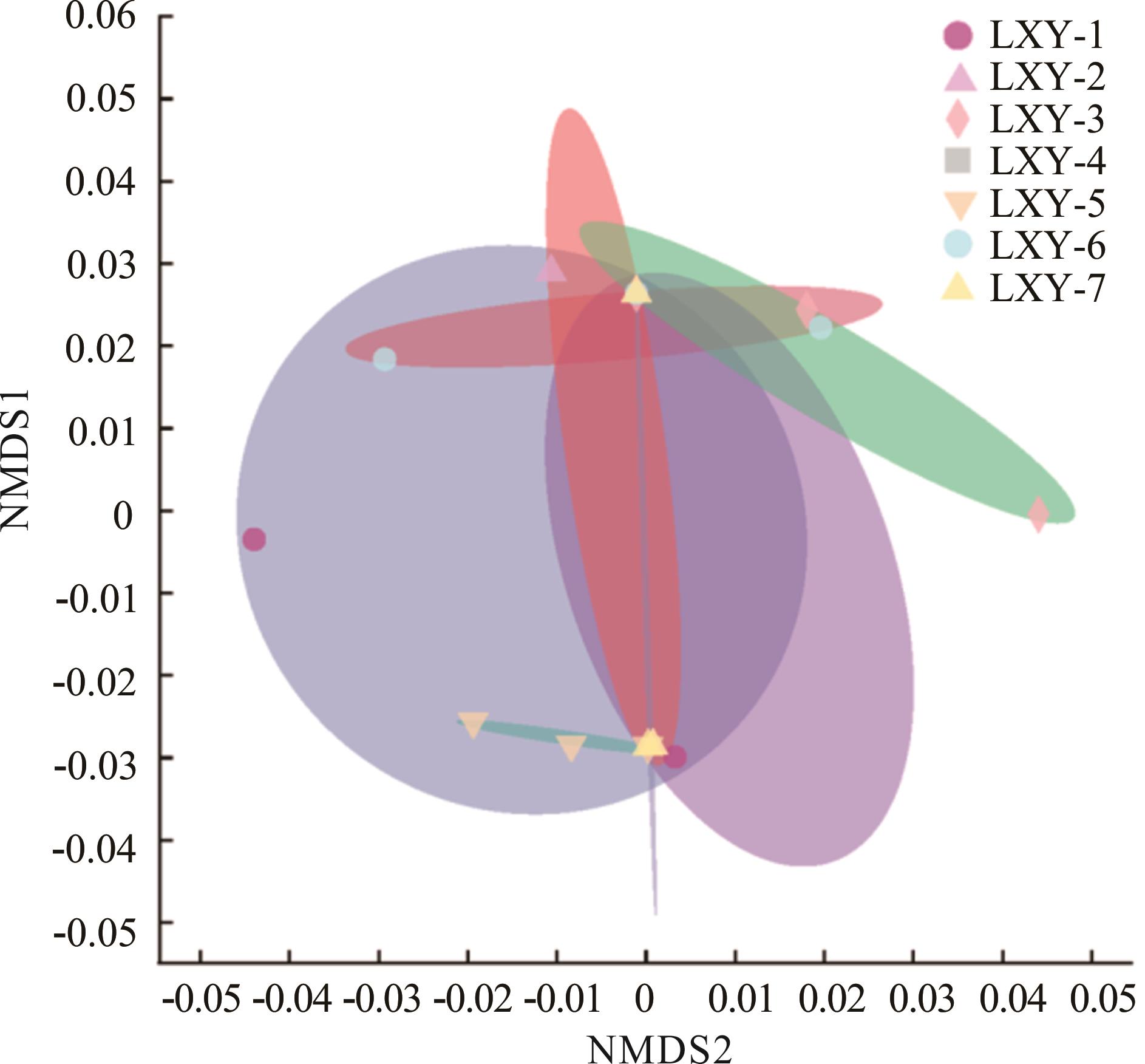
Fig.3 NMDS of seed-borne bacterial community structure in 7 oat cultivars

Fig.4 Linear discriminant analysis of oat seed-borne bacteria

Fig.5 Heat map of seed-borne bacteria KEGG functional diversity in different oat varieties
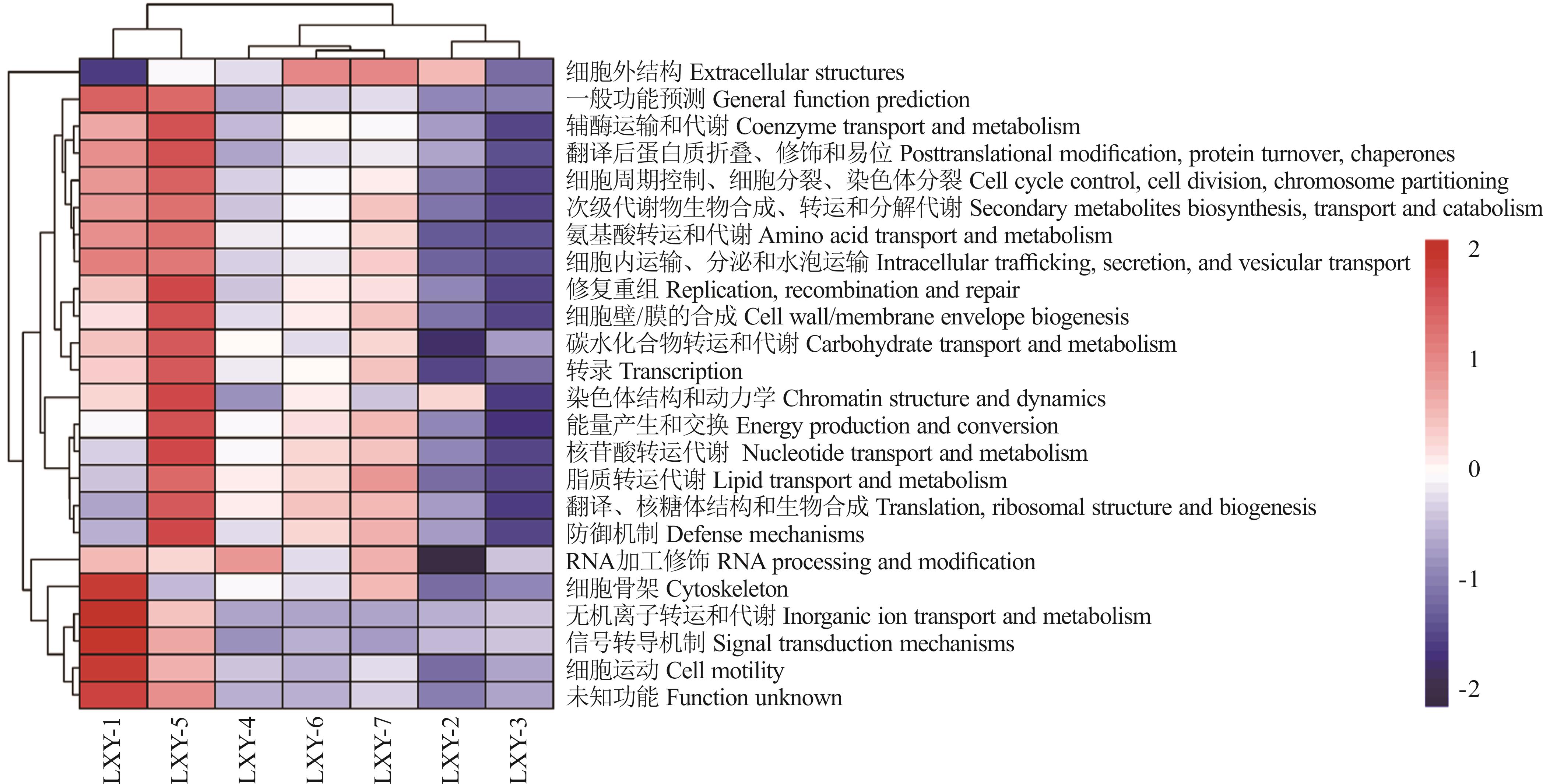
Fig.6 Heat map of seed-borne bacteria COG functional diversity in different oat varieties
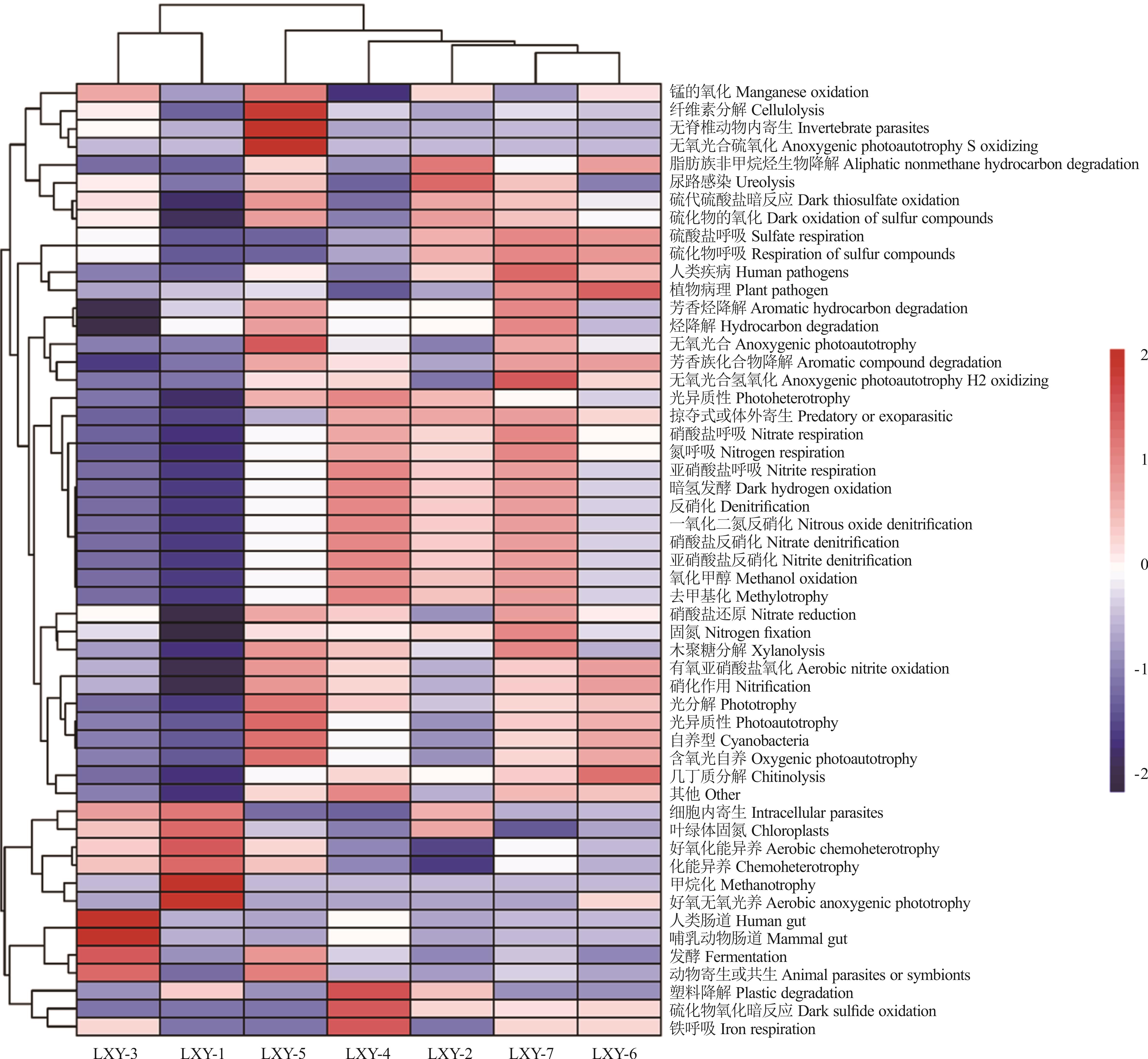
Fig.7 Heat map of seed-borne bacteria FAPROTAX functional diversity in different oat varieties
| 1 | Grum M, Camloh M, Rudolph K, et al. Elimination of bean seed-borne bacteria by thermotherapy and meristem culture. Plant cell, Tissue and Organ Culture, 1998, 52(1): 79-82. |
| 2 | Nan Z B. Alfalfa disease and its comprehensive control system in China. Animal Science and Veterinary Medicine, 2001, 18(4): 81-84. |
| 南志标. 我国的苜蓿病害及其综合防治体系. 动物科学与动物医学, 2001, 18(4): 81-84. | |
| 3 | Gitaitis R, Walcott R. The epidemiology and management of seed-borne bacterial diseases. Annual Review of Phytopathology, 2007, 45(1): 371-397. |
| 4 | Trivedi P, Leach J E, Tringe S G, et al. Plant-microbiome interactions: From community assembly to plant health. Nature Reviews Microbiology, 2020, 18(11): 607-621. |
| 5 | Ahmad M, Dar Z A, Habib M. A review on oat (Avena sativa L.) as a dual-purpose crop. Scientific Research and Essays, 2014, 9(4): 52-59. |
| 6 | Boczkowska M, Tarczyk E. Genetic diversity among polish landraces of common oat (Avena sativa L.). Genetic Resource Crop and Evolution, 2013, 60(7): 2157-2169. |
| 7 | Ren C Z, Yan J T, Dong R, et al. Research progress on oat nutrients, functional properties and related products. Science and Technology of Food Industry, 2022, 43(12): 438-446. |
| 任长忠, 闫金婷, 董锐, 等. 燕麦营养成分、功能特性及其产品的研究进展. 食品工业科技, 2022, 43(12): 438-446. | |
| 8 | Ren C Z, Cui L, Yang C, et al. Establishment and application of high efficient breeding technology system of oat in China. Journal of Agricultural Science and Technology, 2016, 18(1): 1-6. |
| 任长忠, 崔林, 杨才, 等. 我国燕麦高效育种技术体系创建与应用. 中国农业科技导报, 2016, 18(1): 1-6. | |
| 9 | Hu W D, Cao X J, Wu J Y, et al. Effects of fermentation time on fermentation quality and microbial community of oat silage. Feed Research, 2022, 45(17): 106-110. |
| 胡炜东, 曹晓娟, 武俊英, 等. 发酵时间对燕麦青贮发酵品质和微生物群落的影响. 饲料研究, 2022, 45(17): 106-110. | |
| 10 | Chen K X, Li H Y. Research progress of seed endophytes and their application prospect. Chinese Wild Plant Resources, 2021, 40(11): 40-44. |
| 陈柯璇, 李海燕. 植物种子内生菌及其应用前景的研究进展. 中国野生植物资源, 2021, 40(11): 40-44. | |
| 11 | Liu L, Li T Y, Wei H, et al. Effects of a nutrient additive on the density of functional bacteria and the microbial community structure of bioorganic fertilizer. Bioresource Technology, 2014, 172: 328-334. |
| 12 | Mammoudi N, Robeson M S, Castro H F, et al. Microbial community composition and diversity in Caspian Sea sediments. FEMS Microbiology Ecology, 2015, 91(1): 1-11. |
| 13 | Xia X D, Liu X L, Wang Y, et al. Isolation and identification of spoilage organisms in whitefish. Food Science, 2015, 36(21): 175-179. |
| 夏秀东, 刘小莉, 王英, 等. 白鱼腐败细菌的分离与鉴定. 食品科学, 2015, 36(21): 175-179. | |
| 14 | Edgar R. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 2013, 10: 996-998. |
| 15 | Chen K, Chen M, Lin L, et al. Analysis of microbial community characteristics in 9 kinds of chinese herbal pieces based on 16S rRNA high throughput sequencing. Chinese Journal of Medicine Guide, 2022, 28(9): 53-56, 61. |
| 陈柯, 陈敏, 林黎, 等. 基于16S rRNA高通量测序分析9种中药饮片污染微生物群落特征. 中医药导报, 2022, 28(9): 53-56, 61. | |
| 16 | Legendre P, Legendre L. Chapter 6-Withdrawn: Multidimensional qualitative data. Developments in Environmental Modelling, 1998, 20: 207-245. |
| 17 | Wang W, Liu A, Fu W, et al. Tobacco-associated with Methylophilus sp. FP-6 enhances phytoremediation of benzophenone-3 through regulating soil microbial community, increasing photosynthetic capacity and maintaining redox homeostasis of plant. Journal of Hazardous Materials, 2022, 431: 128588. |
| 18 | Langille M G I, Zaneveld J, Caporaso J G, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature Biotechnology, 2013, 31(9): 814-821. |
| 19 | Louca S, Parfrey L W, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science, 2016, 353(6305): 1272-1277. |
| 20 | Shade A, Jacques M A, Barret M. Ecological patterns of seed microbiome diversity, transmission, and assembly. Current Opinion in Microbiology, 2017, 37: 15-22. |
| 21 | Rochefort A, Simonin M, Marais C, et al. Transmission of seed and soil microbiota to seedling. mSystems, 2021, 6(3): e00446-21. |
| 22 | Barret M, Briand M, Bonneau S, et al.Emergence shapes the structure of the seed microbiota. Applied and Environmental Microbiology, 2015, 81(4): 1257-1266. |
| 23 | Chee-Sanford J C, Li M, Davis A S, et al. Symposium do microorganisms influence seed-bank dynamics? Weed Science, 2019, 54(3): 575-587. |
| 24 | Bastias D A, Martinez-Ghersa M A, Ballaré C L, et al. Epichloë fungal endophytes and plant defenses: Not just alkaloids. Trends in Plant Science, 2017, 22(11): 939-948. |
| 25 | Truyens S, Weyens N, Cuypers A, et al. Bacterial seed endophytes: Genera, vertical transmission and interaction with plants. Environmental Microbiology, 2015, 7(1): 40-50. |
| 26 | Kim H, Lee Y H. Spatiotemporal assembly of bacterial and fungal communities of seed-seedling-adult in rice. Frontiers in Microbiology, 2021, 12: 2265. |
| 27 | Lawson C E, Wu S, Bhattacharjee A S, et al. Metabolic network analysis reveals microbial community interactions in anammox granules. Nature Communications, 2017, 8: 15416. |
| 28 | Chen Z B, Huang L, Xia Z Y, et al. Species diversity characteristics of endophytic bacteria in tobacco at different regions of Yunnan Province. Southwest China Journal of Agricultural Sciences, 2015, 28(2): 857-861. |
| 陈泽斌, 黄丽, 夏振远, 等. 云南不同地区烟草内生细菌多样性特征. 西南农业学报, 2015, 28(2): 857-861. | |
| 29 | Qessaoui R, Bouharroud R, Furze J N, et al. Applications of new rhizobacteria Pseudomonas isolates in agroecology via fundamental processes complementing plant growth. Scientific Reports, 2019, 9: 12832. |
| 30 | Matsumoto H, Fan X, Wang Y, et al. Bacterial seed endophyte shapes disease resistance in rice. Nature Plants, 2021, 7(1): 60-72. |
| 31 | Gao M L, Dong Y M, Zhang Z, et al. Effect of dibutyl phthalate on microbial function diversity and enzyme activity in wheat rhizosphere and non-rhizosphere soils. Environmental Pollution, 2020, 265 part B: 114800. |
| 32 | Zhang Z F, Shi S L. Identification and biological attributes of seed-borne bacteria isolated from lucerne cv. Gannong No.3. Acta Prataculturae Sinica, 2018, 27(1): 152-160. |
| 张振粉, 师尚礼.甘农三号紫花苜蓿种带细菌的生物功能分析及鉴定. 草业学报, 2018, 27(1): 152-160. | |
| 33 | Nannipieri P, Ascher J, Ceccherini M T, et al. Microbial diversity and soil functions. European Journal of Soil Science, 2017, 68: 12-26. |
| 34 | Guo H, Mao Z Q, Liu X L. Research progress of interaction between plant and microorganism. Chinese Agricultural Science Bulletin, 2011, 27(9): 28-33. |
| 国辉, 毛志泉, 刘训理. 植物与微生物互作的研究进展. 中国农学通报, 2011, 27(9): 28-33. | |
| 35 | Allu S, Kumar N P, Audipudi A V. Isolation, biochemical and PGP characterization of endophytic Pseudomonas aeruginosa isolated from chilli red fruit antagonistic against chilli anthracnose disease. International Journal of Current Microbiology and Applied Sciences, 2014, 3(2): 318-329. |
| 36 | Zhang R. Identification and detection of seed-born bacterium in rice. Changsha: Hunan Agricultural University, 2014. |
| 张蕊. 水稻种传细菌的检测及鉴定. 长沙: 湖南农业大学, 2014. | |
| 37 | Bastias D A, Bustos L B, Jauregui R, et al. Epichloë fungal endophytes influence seed-associated bacterial communities. Frontiers in Microbiology, 2022, 12: 795354. |
| 38 | Gao G Y. Effects of Epichloië endophyte on microbiomics in seed and seed metabolites of Achnatherum inebriant. Lanzhou: Lanzhou University, 2022. |
| 高国玉. 内生真菌对醉马草种带微生物及种子代谢物的影响. 兰州: 兰州大学, 2022. | |
| 39 | Liang Y T, Xiao X, Nuccio E E, et al. Differentiation strategies of soil rare and abundant microbial taxa in response to changing climatic regimes. Environmental Microbiology, 2020, 22(4): 1327-1340. |
| 40 | Xu G Q. Diversity of endophytes in Oxytropis glacialis on the Qinghai-Tibet Plateau and its adaptability to the environment. Lhasa: Tibet University, 2020. |
| 许国琪. 青藏高原冰川棘豆(Oxytropis glacialis)内生菌多样性及其与环境的适应性研究. 拉萨: 西藏大学, 2020. | |
| 41 | He X H. Dissertation analysis of endophytic microbial community structure of quinoa based on high-throughput sequencing. Chengdu: Chengdu University, 2021. |
| 何小慧. 基于高通量测序的藜麦内生微生物群落结构分析. 成都: 成都大学, 2021. | |
| 42 | Yao L A, Hu Z B, Wang L L, et al. Research development of the relationship between plant endophyte and host. Ecology and Environment, 2010, 19(7): 1750-1754. |
| 姚领爱, 胡之璧, 王莉莉, 等. 植物内生菌与宿主关系研究进展. 生态环境学报, 2010, 19(7): 1750-1754. | |
| 43 | Yan D, Zhang B T, Su A J, et al. Structural variation in the bacterial community associated with airborne particulate matter in Beijing, China, during hazy and nonhazy days. Applied and Environmental Microbiology, 2018, 84(9): e4-e18. |
| 44 | Garcfa-Garcia N, Tamames J, Linz A M, et al. Microdiversity ensures the maintenance of functional microbial communities under changing environmental conditions. The ISME Journal, 2019, 13: 2969-2983. |
| [1] | Qian CHEN, Xiao-yun XU, Jun-cheng WANG, Li-rong YAO, Er-jing SI, Ke YANG, Xiao-ling WEI, Xiao-le MA, Bao-chun LI, Xun-wu SHANG, Ya-xiong MENG, Hua-jun WANG. Identification of a WRKY gene family based on full-length transcriptome sequences and analysis of response patterns under salt stress in Halogeton glomeratus [J]. Acta Prataculturae Sinica, 2022, 31(12): 146-157. |
| [2] | Xin MA, Zhu-zhu LUO, Yao-quan ZHANG, Jia-he LIU, Yi-ning NIU, Li-qun CAI. Distribution characteristics and ecological function predictions of soil bacterial communities in rainfed alfalfa fields on the Loess Plateau [J]. Acta Prataculturae Sinica, 2021, 30(3): 54-67. |
| [3] | SHI Jing-Ang, ZHANG Bing, XIAO Xiao-Lin, MA Jing-Jing, YANG Xiang-Yang, LIU Jian-Xiu. Genome-wide identification and characterization of the cinnamyl alcohol dehydrogenase gene family in Zoysia japonica [J]. Acta Prataculturae Sinica, 2017, 26(6): 111-119. |
| [4] | CHEN Ting-ting,YANG Qing-chuan, ZHANG Xin-quan, KANG Jun-mei, DING Wang, ZHANG Tie-jun. Bioinformatics and expression analyses of ethylene response factor genes in Medicago [J]. Acta Prataculturae Sinica, 2012, 21(6): 166-174. |
| Viewed | ||||||
|
Full text |
|
|||||
|
Abstract |
|
|||||