

ISSN 1004-5759 CN 62-1105/S
草业学报 ›› 2025, Vol. 34 ›› Issue (9): 147-161.DOI: 10.11686/cyxb2024418
• 研究论文 • 上一篇
张志鹏( ), 蒋庆雪, 周昕越, 苗童, 唐俊, 仪登霞, 王学敏(), 马琳()
), 蒋庆雪, 周昕越, 苗童, 唐俊, 仪登霞, 王学敏(), 马琳()
收稿日期:2024-10-24
修回日期:2024-12-02
出版日期:2025-09-20
发布日期:2025-07-02
通讯作者:
王学敏,马琳
作者简介:malin@caas.cn基金资助:
Zhi-peng ZHANG(), Qing-xue JIANG, Xin-yue ZHOU, Tong MIAO, Jun TANG, Deng-xia YI, Xue-min WANG(), Lin MA()
Received:2024-10-24
Revised:2024-12-02
Online:2025-09-20
Published:2025-07-02
Contact:
Xue-min WANG,Lin MA
摘要:
饲用燕麦是一种高产、优质且抗逆性强的饲草,在我国饲草产业的发展中占据重要地位。本研究分别对饲用燕麦高株高(编号972)与低株高(编号1289)的极端材料取穗下茎节和节间组织进行高通量转录组测序(RNA-Seq)和蛋白组定量分析,筛选差异表达基因(differentially expressed genes, DEGs)和差异表达蛋白(differentially expressed proteins, DEPs)。转录组分析筛选到22762个差异表达基因;蛋白组分析获得3934个差异表达蛋白;进一步的联合分析发现1147个差异表达基因(蛋白)重叠出现于转录组及蛋白组分析中。通过对重叠基因(蛋白)进行GO功能富集分析和KEGG信号通路分析,发现很多基因被显著富集到与细胞生长、代谢和细胞壁形成的通路上。进一步结合转录因子分析,筛选到10个饲用燕麦株高性状相关的候选基因。对候选基因进行qRT-PCR验证,得到了测序结果的可靠性。此外,对10个候选基因的组织表达特异性分析发现候选基因在燕麦茎、茎节中均有较高表达,而在其他组织中表达量较低,表明以上候选基因可能参与饲用燕麦的株高发育过程。综上,通过转录组与蛋白组联合分析,结合差异基因功能注释及转录因子分析,筛选到10个饲用燕麦株高性状相关的候选基因。以上候选基因主要通过调控细胞生长、代谢和细胞壁发育参与饲用燕麦株高性状的形成。本研究为进一步探究饲用燕麦株高性状形成的分子机制奠定了基础,并为今后饲用燕麦株高性状的生物育种提供了关键候选基因。
张志鹏, 蒋庆雪, 周昕越, 苗童, 唐俊, 仪登霞, 王学敏, 马琳. 转录组和蛋白组联合筛选饲用燕麦株高性状候选基因[J]. 草业学报, 2025, 34(9): 147-161.
Zhi-peng ZHANG, Qing-xue JIANG, Xin-yue ZHOU, Tong MIAO, Jun TANG, Deng-xia YI, Xue-min WANG, Lin MA. Screening of candidate genes for plant height in forage oat (Avena sativa) through combined transcriptome and proteome analysis[J]. Acta Prataculturae Sinica, 2025, 34(9): 147-161.
编号 Code | 名称 Name | 材料来源 Source of material |
|---|---|---|
| 972 | Dookie 10 | 澳大利亚,维多利亚州Victoria, Australia |
| 1289 | X61-7 |
表1 材料具体信息
Table 1 Materials details
编号 Code | 名称 Name | 材料来源 Source of material |
|---|---|---|
| 972 | Dookie 10 | 澳大利亚,维多利亚州Victoria, Australia |
| 1289 | X61-7 |
| 基因ID Gene ID | 正向引物Forward primer (5′-3′) | 反向引物Reverse primer (5′-3′) |
|---|---|---|
| AVESA.00010b.r1.2CG3384710 | CATCACCGCCAACATCACC | GCAGCCTCCACGATCTCC |
| AVESA.00010b.r1.3CG0669440 | AAGAAGTTGGTGGTTATTGG | CTGCTTCCTTACCTCTCC |
| AVESA.00010b.r1.4AG2537620 | CGGACCTACAACCAGAACC | CAGCCCAAAGTGCCTCTC |
| AVESA.00010b.r1.5DG0437330 | CGGTCTTCTTGCGAATGG | CGATGTACTCCACGAAACC |
| AVESA.00010b.r1.3CG2847600 | ATGCTCTTCACCGTCTCC | CTGGTTGATGTAGTCCTTGG |
| AVESA.00010b.r1.4AG1741330 | GCATAGCGAGCAGTGAAGAG | ACACGGAGATCAGCAGAGC |
| AVESA.00010b.r1.2CG1087750 | CTCCTCCTCCTGCTCGTC | GTGCCCTTGAACCTGTGG |
| AVESA.00010b.r1.2DG0384520 | TGTGCTACGGGAGAAACG | AACTTGATGCCGCTATTCG |
| AVESA.00010b.r1.2CG0426050 | ACCTTCACGCTCAACTTCTCC | GCTCTCCACGCAGTTCTCC |
| AVESA.00010b.r1.4AG0885780 | AACTTCCCGTGCTCTGATCC | GTCGTCTCGTCGGCTTCC |
表2 实时荧光定量 PCR引物设计序列
Table 2 Primer sequences of real-time fluorescence quantitative PCR
| 基因ID Gene ID | 正向引物Forward primer (5′-3′) | 反向引物Reverse primer (5′-3′) |
|---|---|---|
| AVESA.00010b.r1.2CG3384710 | CATCACCGCCAACATCACC | GCAGCCTCCACGATCTCC |
| AVESA.00010b.r1.3CG0669440 | AAGAAGTTGGTGGTTATTGG | CTGCTTCCTTACCTCTCC |
| AVESA.00010b.r1.4AG2537620 | CGGACCTACAACCAGAACC | CAGCCCAAAGTGCCTCTC |
| AVESA.00010b.r1.5DG0437330 | CGGTCTTCTTGCGAATGG | CGATGTACTCCACGAAACC |
| AVESA.00010b.r1.3CG2847600 | ATGCTCTTCACCGTCTCC | CTGGTTGATGTAGTCCTTGG |
| AVESA.00010b.r1.4AG1741330 | GCATAGCGAGCAGTGAAGAG | ACACGGAGATCAGCAGAGC |
| AVESA.00010b.r1.2CG1087750 | CTCCTCCTCCTGCTCGTC | GTGCCCTTGAACCTGTGG |
| AVESA.00010b.r1.2DG0384520 | TGTGCTACGGGAGAAACG | AACTTGATGCCGCTATTCG |
| AVESA.00010b.r1.2CG0426050 | ACCTTCACGCTCAACTTCTCC | GCTCTCCACGCAGTTCTCC |
| AVESA.00010b.r1.4AG0885780 | AACTTCCCGTGCTCTGATCC | GTCGTCTCGTCGGCTTCC |

图1 株高极端燕麦材料的表型分析972:高株高种质High plant height germplasm; 1289: 低株高种质Low plant height germplasm. A: 成熟期的植株(标尺=10 cm)Plants at maturity (scale bar=10 cm); C: 节数、各茎节长度和穗长表型(标尺=5 cm)Number of nodes, length of each stem node and spike length phenotype (scale bar=5 cm). **: P<0.01.
Fig.1 Phenotypic observations of extreme plant height materials
样品名称 Sample | 过滤后数据 Clean reads (M) | 过滤后序列 Clean bases (G) | 碱基质量>30的 占比Q30 (%) | GC含量 GC content (%) | 唯一比对(对比率) Uniquely mapped (mapping ratio, %) | 多方比对(对比率) Multiple mapped (mapping ratio, %) |
|---|---|---|---|---|---|---|
| H-1 | 43.09 | 6.11 | 94.99 | 53.21 | 83.39 | 14.16 |
| H-2 | 43.05 | 6.13 | 95.07 | 52.98 | 83.80 | 14.22 |
| H-3 | 43.08 | 6.13 | 94.84 | 53.08 | 83.92 | 14.09 |
| L-1 | 43.13 | 6.33 | 94.06 | 51.53 | 85.83 | 11.67 |
| L-2 | 44.62 | 6.57 | 94.58 | 51.38 | 85.79 | 11.62 |
| L-3 | 42.36 | 6.23 | 93.99 | 51.16 | 85.83 | 11.60 |
表3 转录组测序数据质控分析
Table 3 Quality test of transcriptome sequencing data
样品名称 Sample | 过滤后数据 Clean reads (M) | 过滤后序列 Clean bases (G) | 碱基质量>30的 占比Q30 (%) | GC含量 GC content (%) | 唯一比对(对比率) Uniquely mapped (mapping ratio, %) | 多方比对(对比率) Multiple mapped (mapping ratio, %) |
|---|---|---|---|---|---|---|
| H-1 | 43.09 | 6.11 | 94.99 | 53.21 | 83.39 | 14.16 |
| H-2 | 43.05 | 6.13 | 95.07 | 52.98 | 83.80 | 14.22 |
| H-3 | 43.08 | 6.13 | 94.84 | 53.08 | 83.92 | 14.09 |
| L-1 | 43.13 | 6.33 | 94.06 | 51.53 | 85.83 | 11.67 |
| L-2 | 44.62 | 6.57 | 94.58 | 51.38 | 85.79 | 11.62 |
| L-3 | 42.36 | 6.23 | 93.99 | 51.16 | 85.83 | 11.60 |

图2 蛋白质鉴定基本信息A: 肽段长度范围分布Distribution of peptide length ranges; B: 蛋白质分子量的分布Distribution of protein molecular weights.
Fig.2 Basic information for protein identification

图3 极端材料差异表达基因和差异表达蛋白上调和下调基因数目
Fig.3 Number of differentially expressed genes and differentially expressed proteins up- and down-regulated genes in extreme materials

图4 转录组和蛋白组关联分析A: 差异表达转录组与蛋白组Venn图Venn diagram of differentially expressed transcriptome and proteome; B: 转录组与蛋白组表达相关性分析Transcriptome and proteome expression correlation analysis; C: 差异表达转录组与蛋白组数目统计Statistics of the number of differentially expressed transcriptome and proteome.
Fig.4 Transcriptome and proteome association analysis

图5 相同变化趋势相关因子的GO富集分析A: 转录本与蛋白共同上调的GO富集分析GO enrichment analysis of transcripts and proteins jointly up-regulated; B: 转录本与蛋白共同下调的GO富集分析GO enrichment analysis of transcripts and proteins jointly down-regulated.
Fig.5 GO enrichment analysis of correlators with the same trend change

图6 相同变化趋势相关因子的KEGG通路分析A: 转录本与蛋白共同上调的KEGG通路分析Analysis of KEGG pathway jointly up-regulated by transcripts and proteins; B: 转录本与蛋白共同下调的KEGG通路分析Analysis of KEGG pathway jointly down-regulated by transcripts and proteins.
Fig.6 KEGG pathway analysis of correlators with the same change trend

图7 不同变化趋势相关因子的GO富集分析A: 转录本下调、蛋白上调的GO富集分析GO enrichment analysis of transcript down-regulation and protein up-regulation; B: 转录本上调、蛋白下调的GO富集分析GO enrichment analysis of transcript up-regulation and protein down-regulation.
Fig.7 GO enrichment analysis of correlators with different trends

图8 不同变化趋势相关因子的KEGG通路分析A: 转录本下调、蛋白上调的KEGG通路分析Analysis of KEGG pathway with transcript down-regulation and protein up-regulation; B: 转录本上调、蛋白下调的KEGG通路分析Analysis of KEGG pathway with transcript up-regulation and protein down-regulation.
Fig.8 KEGG pathway analysis of different trend correlators
变化趋势 | 转录因子家族 Transcription factor family | 数量 Number |
|---|---|---|
| 转录本与蛋白共同上调Transcripts and proteins jointly up-regulated | Nin-like | 1 |
| 转录本与蛋白共同下调Transcripts and proteins jointly down-regulated | Trihelix | 3 |
| FAR1 | 1 | |
| ERF | 1 | |
| 转录本下调、蛋白上调Transcript down-regulation and protein up-regulation | TCP | 1 |
| bHLH | 1 | |
| NF | 1 | |
| 转录本上调、蛋白下调Transcript up-regulation and protein down-regulation | Whirly | 1 |
| NAC | 1 | |
| B3 | 1 |
表4 转录因子分类
Table 4 Classification of transcription factors
变化趋势 | 转录因子家族 Transcription factor family | 数量 Number |
|---|---|---|
| 转录本与蛋白共同上调Transcripts and proteins jointly up-regulated | Nin-like | 1 |
| 转录本与蛋白共同下调Transcripts and proteins jointly down-regulated | Trihelix | 3 |
| FAR1 | 1 | |
| ERF | 1 | |
| 转录本下调、蛋白上调Transcript down-regulation and protein up-regulation | TCP | 1 |
| bHLH | 1 | |
| NF | 1 | |
| 转录本上调、蛋白下调Transcript up-regulation and protein down-regulation | Whirly | 1 |
| NAC | 1 | |
| B3 | 1 |
基因ID Gene ID | 注释 Exegesis | 基因差异倍数Gene fold change | 趋势 Tendencies | 蛋白差异倍数Protein fold change | 趋势 Tendencies |
|---|---|---|---|---|---|
| AVESA.00010b.r1.2CG3384710 | BHLH结构域蛋白质BHLH domain-containing protein | 2.27 | 下调Down | 1.92 | 下调Down |
| AVESA.00010b.r1.3CG0669440 | GIDA葡萄糖抑制分裂蛋白GIDA glucose inhibited division protein | 4.41 | 上调Up | 1.31 | 上调Up |
| AVESA.00010b.r1.4AG2537620 | 糖苷水解酶家族17 Glycosyl hydrolases family 17 | 4.14 | 上调Up | 2.18 | 上调Up |
| AVESA.00010b.r1.5DG0437330 | 未定性蛋白Uncharacterized protein | 4.85 | 下调Down | 1.20 | 上调Up |
| AVESA.00010b.r1.3CG2847600 | 色氨酸氨基转移酶相关蛋白Tryptophan aminotransferase related protein | 5.65 | 下调Down | 3.59 | 下调Down |
| AVESA.00010b.r1.4AG1741330 | 过氧化物酶Peroxidase | 4.69 | 上调Up | 2.50 | 上调Up |
| AVESA.00010b.r1.2CG1087750 | γ-硫堇家族γ-thionin family | 3.70 | 下调Down | 1.28 | 上调Up |
| AVESA.00010b.r1.2DG0384520 | 糖基水解酶家族17 Glycosyl hydrolases family 17 | 4.70 | 上调Up | 1.85 | 上调Up |
| AVESA.00010b.r1.2CG0426050 | 谷氨酸脱羧酶Glutamate decarboxylase | 3.02 | 下调Down | 1.25 | 上调Up |
| AVESA.00010b.r1.4AG0885780 | 乙烯反应性转录因子ERF109 Ethylene-responsive transcription factor ERF109 | 9.04 | 下调Down | 1.89 | 上调Up |
表5 候选基因
Table 5 The candidate genes
基因ID Gene ID | 注释 Exegesis | 基因差异倍数Gene fold change | 趋势 Tendencies | 蛋白差异倍数Protein fold change | 趋势 Tendencies |
|---|---|---|---|---|---|
| AVESA.00010b.r1.2CG3384710 | BHLH结构域蛋白质BHLH domain-containing protein | 2.27 | 下调Down | 1.92 | 下调Down |
| AVESA.00010b.r1.3CG0669440 | GIDA葡萄糖抑制分裂蛋白GIDA glucose inhibited division protein | 4.41 | 上调Up | 1.31 | 上调Up |
| AVESA.00010b.r1.4AG2537620 | 糖苷水解酶家族17 Glycosyl hydrolases family 17 | 4.14 | 上调Up | 2.18 | 上调Up |
| AVESA.00010b.r1.5DG0437330 | 未定性蛋白Uncharacterized protein | 4.85 | 下调Down | 1.20 | 上调Up |
| AVESA.00010b.r1.3CG2847600 | 色氨酸氨基转移酶相关蛋白Tryptophan aminotransferase related protein | 5.65 | 下调Down | 3.59 | 下调Down |
| AVESA.00010b.r1.4AG1741330 | 过氧化物酶Peroxidase | 4.69 | 上调Up | 2.50 | 上调Up |
| AVESA.00010b.r1.2CG1087750 | γ-硫堇家族γ-thionin family | 3.70 | 下调Down | 1.28 | 上调Up |
| AVESA.00010b.r1.2DG0384520 | 糖基水解酶家族17 Glycosyl hydrolases family 17 | 4.70 | 上调Up | 1.85 | 上调Up |
| AVESA.00010b.r1.2CG0426050 | 谷氨酸脱羧酶Glutamate decarboxylase | 3.02 | 下调Down | 1.25 | 上调Up |
| AVESA.00010b.r1.4AG0885780 | 乙烯反应性转录因子ERF109 Ethylene-responsive transcription factor ERF109 | 9.04 | 下调Down | 1.89 | 上调Up |
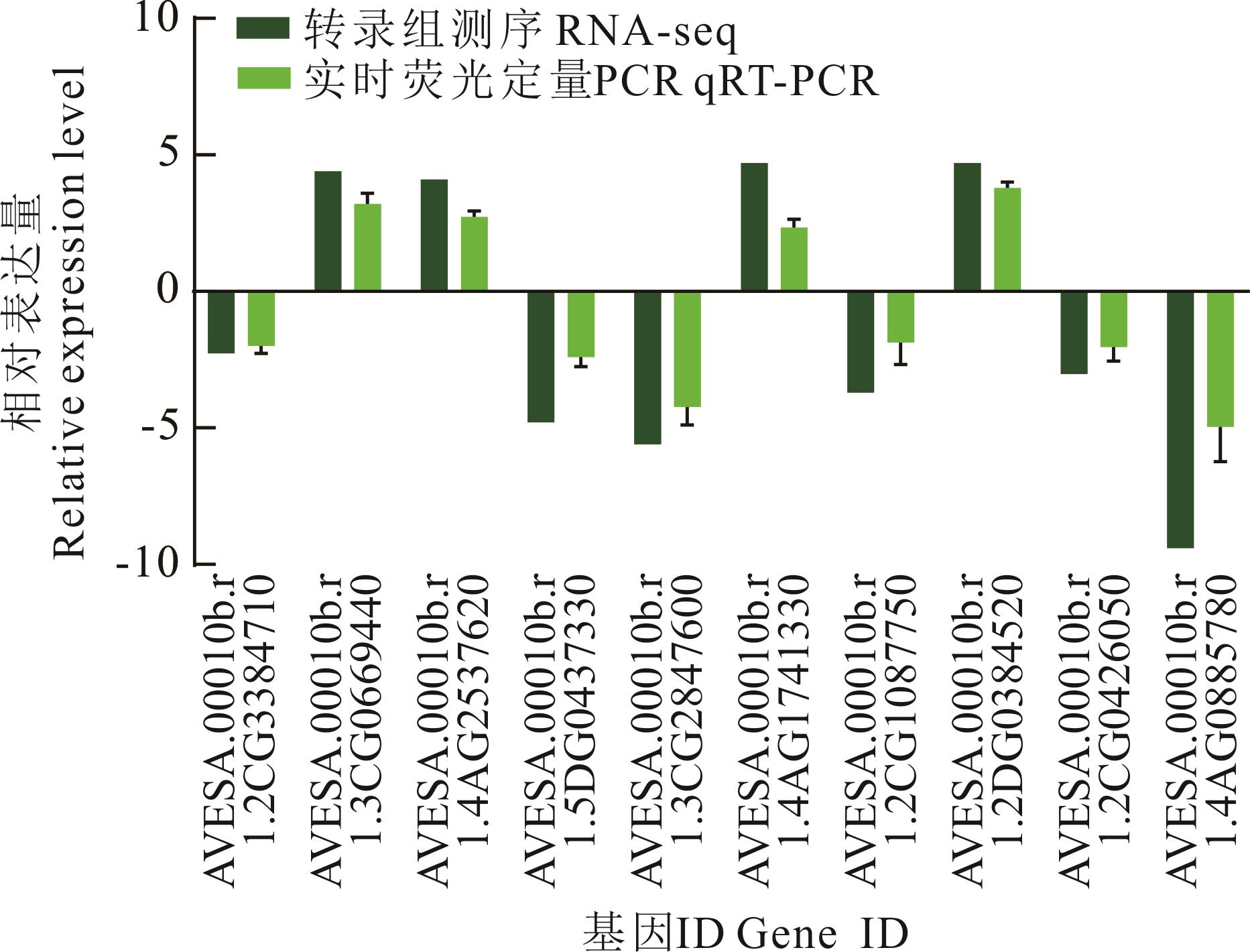
图9 转录组数据的qRT-PCR验证
Fig.9 qRT-PCR validation of transcriptome data

图10 候选基因的组织特异性表达A: 苗期根Seedling stage root; B: 苗期地上Seedling stage aboveground; C: 拔节期根Shooting stage root; D: 拔节期茎Shooting stage stem; E: 拔节期叶Shooting stage leaf; F: 拔节期茎节Shooting stage stem nodes; G: 孕穗期根Booting stage root; H: 孕穗期茎Booting stage stem; I: 孕穗期叶Booting stage leaf; J: 孕穗期茎节Booting stage stem nodes; K: 孕穗期穗Booting stage spike; L: 抽穗期根 Heading stage root; M: 抽穗期茎Heading stage stem; N: 抽穗期叶Heading stage leaf; O: 抽穗期茎节Heading stage stem nodes; P: 抽穗期穗Heading stage spike.
Fig.10 Tissue-specific expression of candidate genes
| [1] | Liu W H, Jia Z F, Liang G L. The current situation, problems and suggestions for the development of China’s oat feed industry. Qinghai Science and Technology, 2020, 27(3): 82-85. |
| 刘文辉, 贾志锋, 梁国玲. 我国饲用燕麦产业发展现状及存在的问题和建议. 青海科技, 2020, 27(3): 82-85. | |
| [2] | Ye X L, Gan Z, Wan Y, et al. Advances and perspectives in forage oat breeding. Acta Prataculturae Sinica, 2023, 32(2): 160-177. |
| 叶雪玲, 甘圳, 万燕, 等. 饲用燕麦育种研究进展与展望. 草业学报, 2023, 32(2): 160-177. | |
| [3] | Chen X, Wu B, Zhang Z W. Evaluation of adaptability and stability for important agronomic traits of oat (Avena spp.) germplasm resources. Journal of Plant Genetic Resources, 2016, 17(4): 577-585. |
| 陈新, 吴斌, 张宗文. 燕麦种质资源重要农艺性状适应性和稳定性评价. 植物遗传资源学报, 2016, 17(4): 577-585. | |
| [4] | Milach S, Rines H W, Phillips R L, et al. Inheritance of a new dwarfing gene in oat. Crop Science, 1998, 38(2): 356-360. |
| [5] | Brown P D, Mckenzie R, Mikaelsen K. Agronomic, genetic, and cytologic evaluation of a vigorous new semidwarf oat. Crop Science, 1980, 20(3): 303-306. |
| [6] | Xu Y H. Fine mapping of oat dwarfing gene Dw6. Chengdu: Sichuan Agricultural University, 2023. |
| 徐颖红. 燕麦矮秆基因Dw6的精细定位. 成都: 四川农业大学, 2023. | |
| [7] | Milach S C K, Rines H W, Phillips R L. Molecular genetic mapping of dwarfing genes in oat. Theoretical & Applied Genetics, 1997, 95(5/6): 783-790. |
| [8] | Morikawa T. Genetic analysis on dwarfness of wild oat, Avena fatua. The Janpanese Journal of Genetics, 1989, 64: 363-371. |
| [9] | Zhang X B. Map-based cloning and functional analysis of leaf shape gene CFL2 and plant height gene SD38 in rice. Chongqing: Southwest University, 2021. |
| 张孝波. 水稻叶形基因CFL2和株高基因SD38的图位克隆与功能分析. 重庆: 西南大学, 2021. | |
| [10] | Yamamuro C, Ihara Y, Wu X, et al. Loss of function of a rice brassinosteroid insensitive1 homolog prevents internode elongation and bending of the lamina joint. The Plant Cell, 2000, 12(9): 1591-1605. |
| [11] | Zhang J, Liu X, Li S, et al. The rice semi-dwarf mutant sd37, caused by a mutation in CYP96B4, plays an important role in the fine-tuning of plant growth. PLoS One, 2014, 9(2): e88068. |
| [12] | Schefe J H, Lehmann K E, Buschmann I R, et al. Quantitative real-time RT-PCR data analysis: Current concepts and the novel “gene expression’s CT difference” formula. Journal of Molecular Medicine, 2006, 84(11): 901-910. |
| [13] | Yang F, Ye R, Ma C, et al. Toxicity evaluation, toxin screening and its intervention of the jellyfish Phacellophora camtschatica based on a combined transcriptome-proteome analysis. Ecotoxicology and Environmental Safety, 2022, 46(6): 1-12. |
| [14] | Luo J. Brassica napus L. dwarf traits proteome and transcriptome joint analysis. Guiyang: Guizhou Normal University, 2021. |
| 罗京. 甘蓝型油菜矮化性状的蛋白质组和转录组联合分析. 贵阳: 贵州师范大学, 2021. | |
| [15] | Li H, He X W, Gao Y F, et al. Integrative analysis of transcriptome, proteome, and phosphoproteome reveals potential roles of photosynthesis antenna proteins in response to brassinosteroids signaling in maize. Plants, 2023, 12(6): 1290-1307. |
| [16] | Lin J, Zheng X, Xia J, et al. Integrative analysis of the transcriptome and proteome reveals the molecular responses of tobacco to boron deficiency. BMC Plant Biology, 2024, 24(1): 689-707. |
| [17] | Wang Y J, Lu W J, Zhao J, et al. Transcriptome dynamics of dominant maize dwarf Dwarf11 (D11) revealed by RNA-seq and co-expression analysis. Plant Molecular Biology Reporter, 2017, 35(3): 355-365. |
| [18] | Sui J M. Fine mapping of one semidwarf gene sdt3 and candidate-gene screening and functional analysis of the other semidwarf gene sdg in rice (Oryza sativa L.) . Yangzhou: Yangzhou University, 2006. |
| 隋炯明. 水稻半矮秆基因sdt3的精细定位和sdg的克隆与功能分析. 扬州: 扬州大学, 2006. | |
| [19] | Gong Y S, Wei S H, Peng Z S, et al. Genetic study on plant height and its components, partial yield traits in durum wheat ‘ANW16F’. Southwest China Journal of Agricultural Sciences, 2021, 34(2): 229-235. |
| 龚胤书, 魏淑红, 彭正松, 等. 硬粒小麦ANW16F株高及构成因子与部分产量性状遗传研究. 西南农业学报, 2021, 34(2): 229-235. | |
| [20] | Wu M Y. Genetic analysis and gene mapping of three (semi-) dwarf genes in rice. Beijing: Chinese Academy of Agricultural Sciences, 2020. |
| 吴明月. 三个(半)矮秆水稻基因的遗传分析和基因定位. 北京: 中国农业科学院, 2020. | |
| [21] | Lv H K, Zhang J, Wang T Y, et al. The maize d2003, a novel allele of VP8, is required for maize internode elongation. Plant Molecular Biology, 2014, 84(3): 243-257. |
| [22] | Peng Z, Li X, Yang Z, et al. A new reduced height gene found in the tetraploid semi-dwarf wheat landrace Aiganfanmai. Genetics and Molecular Research, 2011, 10(4): 2349-2357. |
| [23] | Song J, Li L, Liu B Y, et al. Fine mapping of reduced height locus RHT26 in common wheat. Theoretical and Applied Genetics, 2023, 136(3): 62-72. |
| [24] | Chen L, Yang Y, Mishina K, et al. RNA-seq analysis of the peduncle development of Rht12 dwarf plants and primary mapping of Rht12 in common wheat. Cereal Research Communications, 2020, 48(2): 1-9. |
| [25] | Shan Q Q. Analysis of allelic variaions of wheat plant height regulation genes Rht-1 and GID1 and their interaction mechansim. Zhengzhou: Henan Agricultural University, 2018. |
| 单强强. 小麦株高调控基因Rht-1和GID1的等位变异分析及其编码蛋白的互作机制. 郑州: 河南农业大学, 2018. | |
| [26] | Wang H M. Characterization of the expression patterns of tomato SlGH3.2 and its potential functions in rice plants. Nanjing: Nanjing Agricultural University, 2015. |
| 王慧敏. 番茄SlGH3.2的表达特征及在水稻中的功能研究分析. 南京: 南京农业大学, 2015. | |
| [27] | Ai G. Functional dissection of SlGH3-15 in tomato. Wuhan: Huazhong Agricultural University, 2017. |
| 艾国. 番茄SlGH3-15基因的功能解析. 武汉: 华中农业大学, 2017. | |
| [28] | Kazuhito A, Fumio T. C-terminal extension of rice glutamate decarboxylase (OsGAD2) functions as an autoinhibitory domain and overexpression of a truncated mutant results in the accumulation of extremely high levels of GABA in plant cells. Journal of Experimental Botany, 2007, 58(10): 2699-2707. |
| [29] | Jia Y T. Function analysis of transcription factor bHLH146 in Arabidopsis thaliana. Changchun: Jilin University, 2022. |
| 贾雨彤. 拟南芥转录因子bHLH146的功能研究. 长春: 吉林大学, 2022. | |
| [30] | Qi W W, Sun F, Wang Q J, et al. Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiology, 2011, 157(1): 216-228. |
| [31] | Ma Z M, Wu T, Huang K, et al. A novel AP2/ERF transcription factor, OsRPH1, negatively regulates plant height in rice. Frontiers in Plant Science, 2020, 11(13): 709-724. |
| [1] | 富贵, 刘玉萍, 苏旭, 曲荣举, 才让扎西. 扇穗茅全长转录组SSR特征分析及分子标记开发[J]. 草业学报, 2025, 34(7): 107-119. |
| [2] | 马婷, 陈奋奇, 王勇, 哈雪, 李亚君, 马晖玲. NaCl胁迫下鹰嘴紫云英根系基因差异表达及相关通路分析[J]. 草业学报, 2025, 34(4): 104-123. |
| [3] | 桑瑞娟, 崔超杰, 何云, 张晓霞, 姚晋, 董春阳, 孙浩, 史莹华, 朱晓艳, 李德锋. 豫北地区18个秋播饲用燕麦品种抗倒伏特性及生产性能评价[J]. 草业学报, 2024, 33(8): 74-85. |
| [4] | 李硕, 李培英, 孙宗玖, 李雯. 基于转录组测序的狗牙根抗旱根系关键代谢途径分析[J]. 草业学报, 2024, 33(4): 186-198. |
| [5] | 曾兵, 尚盼盼, 沈秉娜, 王胤晨, 屈明好, 袁扬, 毕磊, 杨兴云, 李文文, 周晓丽, 郑玉倩, 郭文强, 冯彦龙, 曾兵. 淹水胁迫下鸭茅根系基因差异表达及相关通路分析[J]. 草业学报, 2024, 33(2): 93-111. |
| [6] | 余静菠, 张慧丽, 李进, 关皓, 周青平, 陈仕勇. 38份饲用燕麦品种苗期磷利用效率综合评价[J]. 草业学报, 2024, 33(11): 161-171. |
| [7] | 李文龙, 李峰, 张仲鹃, 王殿清, 王欢, 靳慧卿, 特木热, 胡志玲, 陶雅. 鄂尔多斯高原北部一年两季燕麦种植模式生产性能评价[J]. 草业学报, 2024, 33(1): 159-168. |
| [8] | 张浩, 胡海英, 李惠霞, 贺海明, 马霜, 马风华, 宋柯辰. 荒漠草原优势植物牛枝子对干旱胁迫的生理响应与转录组分析[J]. 草业学报, 2023, 32(7): 188-205. |
| [9] | 崔婷, 王勇, 马晖玲. 外源IAA作用下草地早熟禾中调控Cd长距离运输的关键基因表达及其代谢通路分析[J]. 草业学报, 2023, 32(6): 146-156. |
| [10] | 杨瑞鑫, 李勇, 蔡小芳, 韩铖星, 郭艳丽. 不同物理形态的开食料对羔羊瘤胃转录组的影响[J]. 草业学报, 2023, 32(5): 159-170. |
| [11] | 尚盼盼, 曾兵, 屈明好, 李明阳, 杨兴云, 郑玉倩, 沈秉娜, 毕磊, 杨成, 曾兵. 红三叶响应淹水胁迫的相关通路及差异表达基因分析[J]. 草业学报, 2023, 32(4): 112-128. |
| [12] | 叶雪玲, 甘圳, 万燕, 向达兵, 邬晓勇, 吴琪, 刘长英, 范昱, 邹亮. 饲用燕麦育种研究进展与展望[J]. 草业学报, 2023, 32(2): 160-177. |
| [13] | 李峰, 李文龙, 李雪, 张仲鹃, 白林坡, 赵雨霏, 陶雅. 黑龙江中南部16个饲用燕麦品种生产性能综合评价[J]. 草业学报, 2023, 32(10): 82-92. |
| [14] | 孙禄娟, 何建军, 汪军成, 姚立蓉, 司二静, 杨轲, 李葆春, 马小乐, 尚勋武, 孟亚雄, 王化俊. 基于全长转录组测序的盐生草SSR标记开发及其遗传多样性分析[J]. 草业学报, 2022, 31(8): 199-210. |
| [15] | 王志恒, 魏玉清, 赵延蓉, 王悦娟. 基于转录组学比较研究甜高粱幼苗响应干旱和盐胁迫的生理特征[J]. 草业学报, 2022, 31(3): 71-84. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||