

ISSN 1004-5759 CN 62-1105/S
草业学报 ›› 2021, Vol. 30 ›› Issue (5): 103-120.DOI: 10.11686/cyxb2020295
赵小强1( ), 钟源1(), 周文期2
), 钟源1(), 周文期2
收稿日期:2020-06-29
修回日期:2020-08-31
出版日期:2021-05-20
发布日期:2021-04-16
通讯作者:
钟源
作者简介:Corresponding author. E-mail: zhongy@gsau.edu.cn基金资助:
Xiao-qiang ZHAO1(), Yuan ZHONG1(), Wen-qi ZHOU2
Received:2020-06-29
Revised:2020-08-31
Online:2021-05-20
Published:2021-04-16
Contact:
Yuan ZHONG
摘要:
玉米叶面积的大小及分布特征不仅影响其光合效率、蒸腾速率,而且与其耐旱性、耐密性、抗倒伏性及产量形成紧密相关。深入剖析不同水旱环境下玉米不同生育时期不同叶位叶面积的分子遗传机理对玉米耐旱高产新品种的选育具有重要意义。本研究以构建的2套F2∶3群体为试材,在8种水分环境下,采用复合区间作图法(CIM)和基于混合线性模型的复合区间作图法(MCIM)对玉米相应叶(V18时期第10片叶、R1时期穗三叶)叶面积进行单环境和多环境联合QTL分析;参考玉米基因组B73 RefGen_v3挖掘稳定表达的QTLs (sQTLs)区间内的候选基因,并对其进行功能分析。结果表明,采用CIM法,单环境下2个生育时期2套F2∶3群体间总共定位到了7个玉米相应叶叶面积QTLs,主要受显性(81.0%)、部分显性(14.3%)和超显性(4.7%)等遗传效应的调控,其中在干旱环境下定位到了5个QTLs。采用MCIM法,在2套F2∶3群体间总共检测到6个相应叶叶面积的联合QTLs,其中1个表现为显著的QTL与环境的互作(QTL×E, Bin 2.08~2.09),1对QTLs (Bin 1.08~1.10与 Bin 2.08~2.09)参与了显著的加性与加性(AA)上位性互作。结合CIM和MCIM法进一步分析在2套F2∶3群体间检测到了6个sQTLs,其分别位于Bin 1.08~1.10、Bin 2.08~2.09、Bin 4.08~4.09、Bin 6.05、Bin 8.03和Bin 10.03处,并在这些sQTLs区间内确定了12个玉米叶发育相关候选基因。采用生物信息学,总共收集了75个玉米叶发育相关候选基因,通过系统进化树分析表明,这些候选基因划分为3大进化分支,且上述检测到的12个候选基因分布于这3大进化分支上。这些结果为系统地解析玉米不同生育时期不同水旱环境下相应叶叶面积的分子遗传机理提供理论依据,检测到的sQTLs可作为叶面积改良的重要染色体区段,检测到的候选基因为其进一步克隆、功能分析及育种应用提供了信息参考。
赵小强, 钟源, 周文期. 不同水分环境下玉米叶面积QTL定位及候选基因分析[J]. 草业学报, 2021, 30(5): 103-120.
Xiao-qiang ZHAO, Yuan ZHONG, Wen-qi ZHOU. QTL mapping and candidate gene analysis of leaf area in maize (Zea mays) under different watering environments[J]. Acta Prataculturae Sinica, 2021, 30(5): 103-120.
| 时期Stage | 环境 Environment | 双亲Parents | F1杂交种 F1 Hybrid (HLT) | F2∶3群体F2∶3 Population (POPLT,H2=59.76%~64.96%) | |||||
|---|---|---|---|---|---|---|---|---|---|
廊黄 Langhuang | TS141 | 均值 Mean | 变幅 Range | 变异系数 CV (%) | 偏度 Skewness | 峰度 Kurtosis | |||
| V18 | E1 | 301.84±16.84C | 642.41±26.20B | 912.05±36.13A | 510.92±49.92 | 291.79~869.50 | 9.77 | -0.624 | -0.147 |
| E2 | 285.20±20.17C | 463.53±21.72B | 668.23±31.11A | 455.85±46.04 | 257.14~826.57 | 10.10 | 0.370 | 0.245 | |
| E3 | 319.29±20.33C | 667.80±24.29B | 964.37±32.28A | 547.90±46.85 | 328.50~895.18 | 8.55 | 0.462 | 0.103 | |
| E4 | 289.95±18.16C | 480.04±22.75B | 763.19±30.40A | 449.26±49.17 | 260.02~831.84 | 10.95 | 0.911 | -0.069 | |
| R1 | E1 | 510.06±30.42C | 937.51±40.46AB | 1191.58±41.62A | 611.86±65.30 | 340.30~1024.12 | 10.67 | 0.273 | 0.515 |
| E2 | 437.83±34.70B | 540.78±43.22AB | 852.82±39.68A | 519.92±69.49 | 309.62~971.75 | 13.37 | 0.519 | -0.722 | |
| E3 | 537.52±39.95C | 1014.05±42.13B | 1389.28±48.94A | 658.65±62.64 | 373.85~1159.13 | 9.51 | 0.070 | 0.116 | |
| E4 | 479.83±40.12c | 601.03±43.77b | 952.71±45.05a | 541.16±68.81 | 359.81~989.07 | 12.72 | -0.114 | 0.531 | |
| 时期Stage | 环境 Environment | 双亲Parents | F1杂交种 F1 Hybrid (HCT) | F2∶3群体F2∶3 Population (POPCT, H2=64.06%~59.53%) | |||||
昌7-2 Chang7-2 | TS141 | 均值 Mean | 变幅 Range | 变异系数CV (%) | 偏度Skewness | 峰度Kurtosis | |||
| V18 | E5 | 321.20±20.17C | 639.20±28.93B | 900.68±41.00A | 475.80±50.13 | 289.03~846.22 | 10.54 | 0.417 | 0.505 |
| E6 | 300.59±23.10c | 491.12±29.56b | 697.30±38.86a | 430.78±48.80 | 248.49~812.16 | 11.33 | 0.084 | 0.622 | |
| E7 | 326.42±24.52C | 687.88±25.11B | 930.63±45.25A | 527.41±53.96 | 319.75~864.51 | 10.23 | -0.336 | -0.916 | |
| E8 | 296.96±26.39C | 519.23±24.15AB | 711.21±40.18A | 452.80±47.24 | 250.58~830.77 | 10.43 | 0.614 | -0.110 | |
| R1 | E5 | 538.37±37.25B | 1015.16±46.02A | 1197.20±47.61A | 585.84±68.90 | 337.95~977.32 | 11.76 | 0.622 | 0.494 |
| E6 | 436.80±39.11c | 643.80±41.05b | 908.57±41.19a | 507.44±63.25 | 315.48~969.04 | 12.46 | -0.191 | -1.002 | |
| E7 | 552.29±32.48B | 1029.93±42.23A | 1013.00±45.37A | 631.42±69.74 | 394.12~1194.79 | 11.04 | -0.438 | 0.389 | |
| E8 | 457.52±36.49c | 671.95±47.80b | 856.19±37.42a | 530.20±68.06 | 347.88~1018.21 | 12.84 | -0.714 | -0.116 | |
表1 亲本、F1杂交种及F2∶3群体玉米相应叶的叶面积表型
Table 1 Performance of leaf area (LA) for corresponding leaves in parents, F1 hybrids, and F2∶3 populations
| 时期Stage | 环境 Environment | 双亲Parents | F1杂交种 F1 Hybrid (HLT) | F2∶3群体F2∶3 Population (POPLT,H2=59.76%~64.96%) | |||||
|---|---|---|---|---|---|---|---|---|---|
廊黄 Langhuang | TS141 | 均值 Mean | 变幅 Range | 变异系数 CV (%) | 偏度 Skewness | 峰度 Kurtosis | |||
| V18 | E1 | 301.84±16.84C | 642.41±26.20B | 912.05±36.13A | 510.92±49.92 | 291.79~869.50 | 9.77 | -0.624 | -0.147 |
| E2 | 285.20±20.17C | 463.53±21.72B | 668.23±31.11A | 455.85±46.04 | 257.14~826.57 | 10.10 | 0.370 | 0.245 | |
| E3 | 319.29±20.33C | 667.80±24.29B | 964.37±32.28A | 547.90±46.85 | 328.50~895.18 | 8.55 | 0.462 | 0.103 | |
| E4 | 289.95±18.16C | 480.04±22.75B | 763.19±30.40A | 449.26±49.17 | 260.02~831.84 | 10.95 | 0.911 | -0.069 | |
| R1 | E1 | 510.06±30.42C | 937.51±40.46AB | 1191.58±41.62A | 611.86±65.30 | 340.30~1024.12 | 10.67 | 0.273 | 0.515 |
| E2 | 437.83±34.70B | 540.78±43.22AB | 852.82±39.68A | 519.92±69.49 | 309.62~971.75 | 13.37 | 0.519 | -0.722 | |
| E3 | 537.52±39.95C | 1014.05±42.13B | 1389.28±48.94A | 658.65±62.64 | 373.85~1159.13 | 9.51 | 0.070 | 0.116 | |
| E4 | 479.83±40.12c | 601.03±43.77b | 952.71±45.05a | 541.16±68.81 | 359.81~989.07 | 12.72 | -0.114 | 0.531 | |
| 时期Stage | 环境 Environment | 双亲Parents | F1杂交种 F1 Hybrid (HCT) | F2∶3群体F2∶3 Population (POPCT, H2=64.06%~59.53%) | |||||
昌7-2 Chang7-2 | TS141 | 均值 Mean | 变幅 Range | 变异系数CV (%) | 偏度Skewness | 峰度Kurtosis | |||
| V18 | E5 | 321.20±20.17C | 639.20±28.93B | 900.68±41.00A | 475.80±50.13 | 289.03~846.22 | 10.54 | 0.417 | 0.505 |
| E6 | 300.59±23.10c | 491.12±29.56b | 697.30±38.86a | 430.78±48.80 | 248.49~812.16 | 11.33 | 0.084 | 0.622 | |
| E7 | 326.42±24.52C | 687.88±25.11B | 930.63±45.25A | 527.41±53.96 | 319.75~864.51 | 10.23 | -0.336 | -0.916 | |
| E8 | 296.96±26.39C | 519.23±24.15AB | 711.21±40.18A | 452.80±47.24 | 250.58~830.77 | 10.43 | 0.614 | -0.110 | |
| R1 | E5 | 538.37±37.25B | 1015.16±46.02A | 1197.20±47.61A | 585.84±68.90 | 337.95~977.32 | 11.76 | 0.622 | 0.494 |
| E6 | 436.80±39.11c | 643.80±41.05b | 908.57±41.19a | 507.44±63.25 | 315.48~969.04 | 12.46 | -0.191 | -1.002 | |
| E7 | 552.29±32.48B | 1029.93±42.23A | 1013.00±45.37A | 631.42±69.74 | 394.12~1194.79 | 11.04 | -0.438 | 0.389 | |
| E8 | 457.52±36.49c | 671.95±47.80b | 856.19±37.42a | 530.20±68.06 | 347.88~1018.21 | 12.84 | -0.714 | -0.116 | |
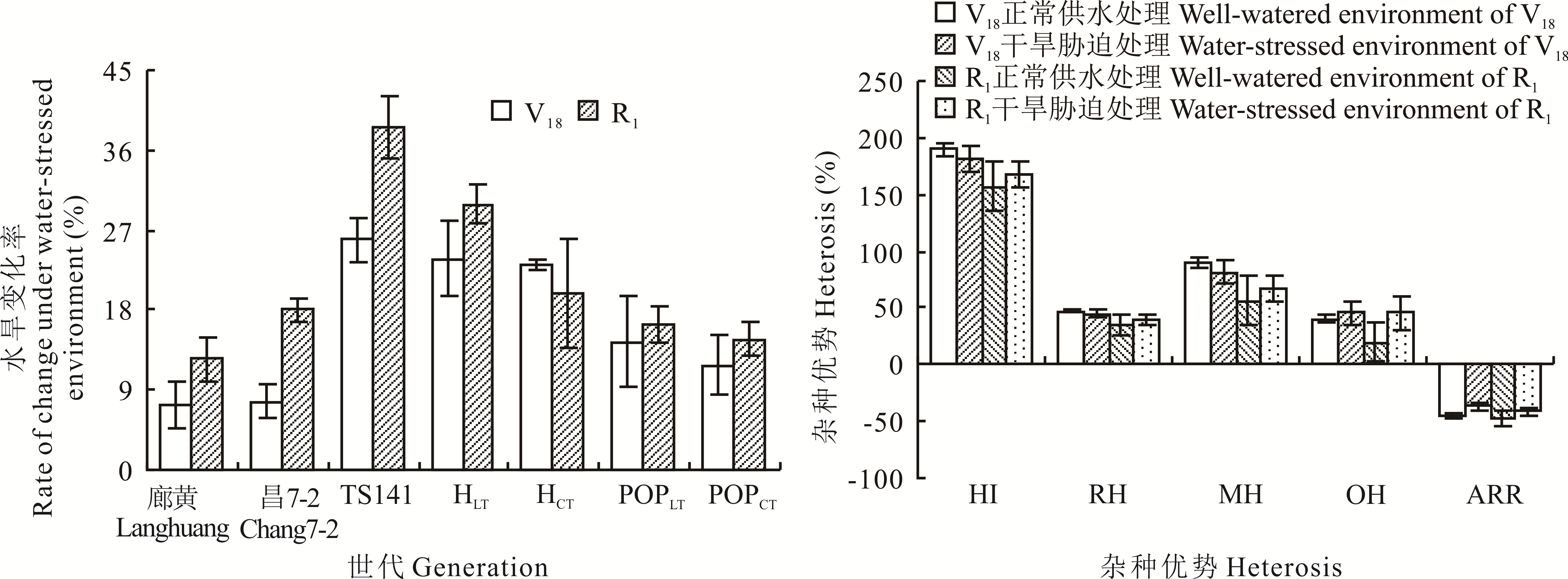
图1 玉米叶面积的水旱变化率和杂种优势分析HI: F1杂种优势指数F1 heterosis index;RH: 相对杂种优势Relative heterosis;MH: 中亲优势Mid-parent heterosis;OH: 超亲优势Over-parent heterosis;ARR: F2∶3优势降低率F2∶3 advantage reduction rate.
Fig.1 Rate of change under water-stressed environment and heterosis analysis of leaf area of maize

图2 F2∶3群体(POPLT和POPCT)相关性状间的相关性分析
Fig.2 Correlative analysis among corresponding traits in F2∶3 populations (POPLT and POPCT)
群体 Population (QTL) | 染色体 Chromosome | 时期 Stage | 环境 Environment | QTL 位置 QTL position | LOD | 遗传效应 Genetic effect | 基因方式 Gene action | 贡献率 Contribution rate (R2 , %) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
图距Graph distance (cM) | 区间 Interval | 加性 Additive | 显性 Dominance | |d/a| | 类型 Type | |||||||
| POPLT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | E2 | 109.6 | mmc0041~phi308707 | 5.47 | 1.58 | 1.62 | 1.03 | D | 8.11 | |
| qLA-Ch.1-1 | 1 | V18 | E3 | 109.6 | mmc0041~phi308707 | 5.90 | -1.51 | 0.93 | 0.62 | PD | 9.79 | |
| qLA-Ch.1-1 | 1 | R1 | E1 | 109.1 | mmc0041~phi308707 | 3.93 | 1.18 | -1.20 | 1.02 | D | 7.00 | |
| qLA-Ch.1-1 | 1 | R1 | E2 | 109.1 | mmc0041~phi308707 | 4.11 | 1.30 | -1.24 | 0.95 | D | 7.13 | |
| qLA-Ch.1-1 | 1 | R1 | E4 | 109.6 | mmc0041~phi308707 | 6.95 | 1.97 | -2.00 | 1.02 | D | 10.40 | |
| qLA-Ch.2-1 | 2 | V18 | E3 | 44.7 | bnlg1909~bnlg1613 | 3.89 | -2.16 | 2.95 | 1.37 | OD | 5.60 | |
| qLA-Ch.4-1 | 4 | V18 | E2 | 180.6 | umc2041~umc2287 | 3.03 | 0.27 | 0.12 | 0.44 | PD | 3.43 | |
| qLA-Ch.4-1 | 4 | R1 | E2 | 180.6 | umc2041~umc2287 | 4.55 | 0.99 | -0.47 | 0.47 | PD | 6.12 | |
| qLA-Ch.8-1 | 8 | V18 | E1 | 45.1 | bnlg1863~umc2075 | 4.00 | 1.19 | -1.25 | 1.05 | D | 6.94 | |
| qLA-Ch.8-1 | 8 | V18 | E2 | 46.0 | bnlg1863~umc2075 | 4.32 | 1.49 | 1.61 | 1.08 | D | 7.22 | |
| qLA-Ch.8-1 | 8 | R1 | E1 | 45.2 | bnlg1863~umc2075 | 3.75 | 1.22 | 1.14 | 0.93 | D | 7.38 | |
| qLA-Ch.8-1 | 8 | R1 | E2 | 46.0 | bnlg1863~umc2075 | 4.49 | 1.37 | 1.51 | 1.10 | D | 8.41 | |
| qLA-Ch.10-1 | 10 | V18 | E3 | 47.2 | bnlg1655~umc1345 | 3.84 | -1.28 | -1.15 | 0.89 | D | 7.51 | |
| qLA-Ch.10-1 | 10 | R1 | E3 | 47.2 | bnlg1655~umc1345 | 6.10 | -1.71 | -1.77 | 1.04 | D | 11.84 | |
| POPCT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | E6 | 156.1 | mmc0041~phi308707 | 3.98 | -1.96 | -2.03 | 1.03 | D | 6.05 | |
| qLA-Ch.1-1 | 1 | R1 | E6 | 156.1 | mmc0041~phi308707 | 5.00 | -1.89 | 2.10 | 1.11 | D | 6.89 | |
| qLA-Ch.1-1 | 1 | R1 | E8 | 156.1 | mmc0041~phi308707 | 4.13 | -2.14 | -2.18 | 1.02 | D | 6.21 | |
| qLA-Ch.6-1 | 6 | V18 | E7 | 90.4 | umc2040~bnlg1174a | 3.95 | -2.00 | -2.22 | 1.11 | D | 6.02 | |
| qLA-Ch.6-1 | 6 | V18 | E8 | 90.4 | umc2040~bnlg1174a | 4.49 | 2.28 | 2.13 | 0.93 | D | 6.49 | |
| qLA-Ch.6-1 | 6 | R1 | E6 | 90.4 | umc2040~bnlg1174a | 7.28 | -1.80 | -1.71 | 0.95 | D | 9.33 | |
| qLA-Ch.6-1 | 6 | R1 | E8 | 90.4 | umc2040~bnlg1174a | 8.00 | -2.82 | -2.49 | 0.88 | D | 11.94 | |
表2 单环境下采用CIM法对F2∶3群体相应叶叶面积QTLs检测
Table 2 QTLs for leaf area were detected in F2∶3 populations by single environment mapping with CIM
群体 Population (QTL) | 染色体 Chromosome | 时期 Stage | 环境 Environment | QTL 位置 QTL position | LOD | 遗传效应 Genetic effect | 基因方式 Gene action | 贡献率 Contribution rate (R2 , %) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
图距Graph distance (cM) | 区间 Interval | 加性 Additive | 显性 Dominance | |d/a| | 类型 Type | |||||||
| POPLT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | E2 | 109.6 | mmc0041~phi308707 | 5.47 | 1.58 | 1.62 | 1.03 | D | 8.11 | |
| qLA-Ch.1-1 | 1 | V18 | E3 | 109.6 | mmc0041~phi308707 | 5.90 | -1.51 | 0.93 | 0.62 | PD | 9.79 | |
| qLA-Ch.1-1 | 1 | R1 | E1 | 109.1 | mmc0041~phi308707 | 3.93 | 1.18 | -1.20 | 1.02 | D | 7.00 | |
| qLA-Ch.1-1 | 1 | R1 | E2 | 109.1 | mmc0041~phi308707 | 4.11 | 1.30 | -1.24 | 0.95 | D | 7.13 | |
| qLA-Ch.1-1 | 1 | R1 | E4 | 109.6 | mmc0041~phi308707 | 6.95 | 1.97 | -2.00 | 1.02 | D | 10.40 | |
| qLA-Ch.2-1 | 2 | V18 | E3 | 44.7 | bnlg1909~bnlg1613 | 3.89 | -2.16 | 2.95 | 1.37 | OD | 5.60 | |
| qLA-Ch.4-1 | 4 | V18 | E2 | 180.6 | umc2041~umc2287 | 3.03 | 0.27 | 0.12 | 0.44 | PD | 3.43 | |
| qLA-Ch.4-1 | 4 | R1 | E2 | 180.6 | umc2041~umc2287 | 4.55 | 0.99 | -0.47 | 0.47 | PD | 6.12 | |
| qLA-Ch.8-1 | 8 | V18 | E1 | 45.1 | bnlg1863~umc2075 | 4.00 | 1.19 | -1.25 | 1.05 | D | 6.94 | |
| qLA-Ch.8-1 | 8 | V18 | E2 | 46.0 | bnlg1863~umc2075 | 4.32 | 1.49 | 1.61 | 1.08 | D | 7.22 | |
| qLA-Ch.8-1 | 8 | R1 | E1 | 45.2 | bnlg1863~umc2075 | 3.75 | 1.22 | 1.14 | 0.93 | D | 7.38 | |
| qLA-Ch.8-1 | 8 | R1 | E2 | 46.0 | bnlg1863~umc2075 | 4.49 | 1.37 | 1.51 | 1.10 | D | 8.41 | |
| qLA-Ch.10-1 | 10 | V18 | E3 | 47.2 | bnlg1655~umc1345 | 3.84 | -1.28 | -1.15 | 0.89 | D | 7.51 | |
| qLA-Ch.10-1 | 10 | R1 | E3 | 47.2 | bnlg1655~umc1345 | 6.10 | -1.71 | -1.77 | 1.04 | D | 11.84 | |
| POPCT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | E6 | 156.1 | mmc0041~phi308707 | 3.98 | -1.96 | -2.03 | 1.03 | D | 6.05 | |
| qLA-Ch.1-1 | 1 | R1 | E6 | 156.1 | mmc0041~phi308707 | 5.00 | -1.89 | 2.10 | 1.11 | D | 6.89 | |
| qLA-Ch.1-1 | 1 | R1 | E8 | 156.1 | mmc0041~phi308707 | 4.13 | -2.14 | -2.18 | 1.02 | D | 6.21 | |
| qLA-Ch.6-1 | 6 | V18 | E7 | 90.4 | umc2040~bnlg1174a | 3.95 | -2.00 | -2.22 | 1.11 | D | 6.02 | |
| qLA-Ch.6-1 | 6 | V18 | E8 | 90.4 | umc2040~bnlg1174a | 4.49 | 2.28 | 2.13 | 0.93 | D | 6.49 | |
| qLA-Ch.6-1 | 6 | R1 | E6 | 90.4 | umc2040~bnlg1174a | 7.28 | -1.80 | -1.71 | 0.95 | D | 9.33 | |
| qLA-Ch.6-1 | 6 | R1 | E8 | 90.4 | umc2040~bnlg1174a | 8.00 | -2.82 | -2.49 | 0.88 | D | 11.94 | |

图3 F2∶3群体(POPLT)检测到的QTLs及上位性效应QTLs在遗传连锁图谱上的分布?●?▲分别表示V18时期检测到的QTL,R1时期检测到的QTL,联合QTL和QTL×E 位点。下同。?●?▲represented the QTL at V18 stage, the QTL at R1 stage, joint QTL, QTL×E. The same below.
Fig.3 Distribution of QTLs and epistatic QTLs on the corresponding linkage genetic map in F2∶3 (POPLT) population

图4 F2∶3群体(POPCT)检测到的QTLs及上位性效应QTLs在遗传连锁图谱上的分布虚线表示加性与加性(AA)上位性互作。Dotted line indicated epistatic interaction with additive by additive (AA) effect.
Fig.4 Distribution of QTLs and epistatic QTLs on the corresponding linkage genetic map in F2∶3 (POPCT) population
群体 Population | 染色体 Chromosome | 时期 Stage | QTL位置QTL position | A | AE1/AE5 | AE2/AE6 | AE3/AE7 | AE4/AE8 | h2 (A) (%) | h2 (AE) (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
图距 Graph distance (cM) | 区间距离 Interval distance (Mb) | 区间 Interval | ||||||||||
| POPLT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | 109.3 | 17.51 | mmc0041~phi308707 | 0.99 | - | - | - | - | 6.58 | - |
| qLA-Ch.1-1 | 1 | R1 | 109.5 | 17.51 | mmc0041~phi308707 | 0.63 | - | - | - | - | 4.84 | - |
| qLA-J2-1 | 2 | R1 | 77.8 | 14.79 | bnlg1233~bnlg1520 | -1.90 | - | 0.66 | - | - | 9.20 | 4.52 |
| qLA-Ch.8-1 | 8 | V18 | 46.0 | 0.39 | bnlg1863~umc2075 | 1.03 | - | - | - | - | 6.68 | - |
| qLA-Ch.8-1 | 8 | R1 | 45.8 | 0.39 | bnlg1863~umc2075 | 0.78 | - | - | - | - | 5.04 | - |
| POPCT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | 156.0 | 17.51 | mmc0041~phi308707 | -0.70 | - | - | - | - | 4.89 | - |
| qLA-Ch.1-1 | 1 | R1 | 156.1 | 17.51 | mmc0041~phi308707 | -0.84 | - | - | - | - | 5.02 | - |
| qLA-J2-1 | 2 | R1 | 116.8 | 14.79 | bnlg1233~bnlg1520 | -1.43 | - | - | - | - | 6.00 | - |
| qLA-Ch.6-1 | 6 | V18 | 90.4 | 11.12 | umc2040~bnlg1174a | -0.66 | - | - | - | - | 4.82 | - |
| qLA-Ch.6-1 | 6 | R1 | 90.4 | 11.12 | umc2040~bnlg1174a | -0.91 | - | - | - | - | 5.09 | - |
表3 多环境下采用MCIM法对F2∶3群体相应叶叶面积联合QTLs及QTL×E分析
Table 3 Joint QTLs and QTL×E for leaf area were detected in F2∶3 populations under multiple environments by MCIM
群体 Population | 染色体 Chromosome | 时期 Stage | QTL位置QTL position | A | AE1/AE5 | AE2/AE6 | AE3/AE7 | AE4/AE8 | h2 (A) (%) | h2 (AE) (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
图距 Graph distance (cM) | 区间距离 Interval distance (Mb) | 区间 Interval | ||||||||||
| POPLT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | 109.3 | 17.51 | mmc0041~phi308707 | 0.99 | - | - | - | - | 6.58 | - |
| qLA-Ch.1-1 | 1 | R1 | 109.5 | 17.51 | mmc0041~phi308707 | 0.63 | - | - | - | - | 4.84 | - |
| qLA-J2-1 | 2 | R1 | 77.8 | 14.79 | bnlg1233~bnlg1520 | -1.90 | - | 0.66 | - | - | 9.20 | 4.52 |
| qLA-Ch.8-1 | 8 | V18 | 46.0 | 0.39 | bnlg1863~umc2075 | 1.03 | - | - | - | - | 6.68 | - |
| qLA-Ch.8-1 | 8 | R1 | 45.8 | 0.39 | bnlg1863~umc2075 | 0.78 | - | - | - | - | 5.04 | - |
| POPCT | ||||||||||||
| qLA-Ch.1-1 | 1 | V18 | 156.0 | 17.51 | mmc0041~phi308707 | -0.70 | - | - | - | - | 4.89 | - |
| qLA-Ch.1-1 | 1 | R1 | 156.1 | 17.51 | mmc0041~phi308707 | -0.84 | - | - | - | - | 5.02 | - |
| qLA-J2-1 | 2 | R1 | 116.8 | 14.79 | bnlg1233~bnlg1520 | -1.43 | - | - | - | - | 6.00 | - |
| qLA-Ch.6-1 | 6 | V18 | 90.4 | 11.12 | umc2040~bnlg1174a | -0.66 | - | - | - | - | 4.82 | - |
| qLA-Ch.6-1 | 6 | R1 | 90.4 | 11.12 | umc2040~bnlg1174a | -0.91 | - | - | - | - | 5.09 | - |
sQTLs区间 sQTLs interval | 基因数量Number of gene | 基因 ID Gene ID | 分子功能 Molecular function | 生物进程 Biological process | 细胞组分 Cell component |
|---|---|---|---|---|---|
mmc0041~ phi308707 | 146 | GRMZM2G014392 | 氧化还原酶活性Oxidoreductase activity | 脱落酸生物合成/氧化还原过程Abscisic acid biosynthetic process/oxidation-reduction process | 叶绿体/质体/叶绿体基质Chloroplast/plastid/chloroplast stroma |
| GRMZM2G073725 | 分子内转移酶/UDP-阿糖吡喃糖酶活性Intramolecular transferase activity/UDP-arabinopyranose mutase activity | 纤维素生物合成过程/细胞壁组织/植物细胞壁组织或生物发生Cellulose biosynthetic process/cell wall organization/plant-type cell wall organization or biogenesis | 组织或生物发生/高尔基体/胞间连丝Cellulose biosynthetic process/plant-type cell wall organization or biogenesis/golgi apparatus/plasmodesma | ||
| GRMZM2G017087 | DNA/RNA/蛋白结合DNA/RNA/protein binding | 转录调节/DNA模板/胞间质转运Regulation of transcription/DNA-templated/plasmodesmata-mediated intercellular transport | 细胞质/胞间连丝/微管细胞骨架Cytoplasm/plasmodesma/microtubule cytoskeleton | ||
bnlg1233~ bnlg1520 | 104 | GRMZM2G140667 | 过氧化物酶活性Peroxidase activity | 氧化应激反应/细胞氧化解毒Response to oxidative stress/cellular oxidant detoxification | 叶绿体/类囊体Chloroplast/thylakoid |
| GRMZM2G005990 | 二氢叶酸还原酶活性/戊二酸合酶活性Dihydrofolate reductase activity/hymidylate synthase activity | dTMP生物合成/单碳代谢dTMP biosynthetic process/one-carbon metabolic process | - | ||
umc2041~ umc22870 | 139 | GRMZM2G094497 | ATP结合ATP binding | ATP水解耦合质子转运/质子转运/ATP代谢过程ATP hydrolysis coupled proton transport/proton transport/ATP metabolic process | 质子运输V型ATP酶/V1结构域Proton-transporting V-type ATPase/V1domain |
| GRMZM2G074122 | 催化活性/苯醇丙酮酸羧化酶活性Catalytic activity/hosphoenolpyruvate carboxylase activity | 三羧酸循环/碳固定Tricarboxylic acid cycle/carbon fixation | - | ||
| GRMZM2G162434 | DNA结合DNA binding | - | - | ||
| GRMZM2G116079 | 金属离子结合 Metalion binding | - | - | ||
umc2040~ bnlg1174a | 24 | GRMZM2G039113 | - | - | 细胞质/细胞骨架Cytoplsm/cytoskeleton |
umc2040~ bnlg1174a | 2 | GRMZM2G119169 | 核糖体结构组成Structural constituent of ribosome | 翻译Translation | 核糖体/核糖体大亚基/细胞内核糖体蛋白复合体Ribosome/large ribosomal subunit/intracellular ribonucleoprotein complex |
umc2040~ bnlg1174a | 48 | GRMZM2G163437 | ATP结合/葡萄糖-1-1磷酸腺苷转移酶活性ATP binding/glucose-1-phosphate adenylytransferase activity | 核糖体生物合成/淀粉生物合成Glycogen biosynthetic process/starch biosynthetic process | 叶绿体/质体Chloroplast/plastid |
表4 叶面积sQTLs区间内候选基因检测及功能分析
Table 4 Candidate genes were detected and corresponding function were analyzed in sQTLs intervals for leaf area
sQTLs区间 sQTLs interval | 基因数量Number of gene | 基因 ID Gene ID | 分子功能 Molecular function | 生物进程 Biological process | 细胞组分 Cell component |
|---|---|---|---|---|---|
mmc0041~ phi308707 | 146 | GRMZM2G014392 | 氧化还原酶活性Oxidoreductase activity | 脱落酸生物合成/氧化还原过程Abscisic acid biosynthetic process/oxidation-reduction process | 叶绿体/质体/叶绿体基质Chloroplast/plastid/chloroplast stroma |
| GRMZM2G073725 | 分子内转移酶/UDP-阿糖吡喃糖酶活性Intramolecular transferase activity/UDP-arabinopyranose mutase activity | 纤维素生物合成过程/细胞壁组织/植物细胞壁组织或生物发生Cellulose biosynthetic process/cell wall organization/plant-type cell wall organization or biogenesis | 组织或生物发生/高尔基体/胞间连丝Cellulose biosynthetic process/plant-type cell wall organization or biogenesis/golgi apparatus/plasmodesma | ||
| GRMZM2G017087 | DNA/RNA/蛋白结合DNA/RNA/protein binding | 转录调节/DNA模板/胞间质转运Regulation of transcription/DNA-templated/plasmodesmata-mediated intercellular transport | 细胞质/胞间连丝/微管细胞骨架Cytoplasm/plasmodesma/microtubule cytoskeleton | ||
bnlg1233~ bnlg1520 | 104 | GRMZM2G140667 | 过氧化物酶活性Peroxidase activity | 氧化应激反应/细胞氧化解毒Response to oxidative stress/cellular oxidant detoxification | 叶绿体/类囊体Chloroplast/thylakoid |
| GRMZM2G005990 | 二氢叶酸还原酶活性/戊二酸合酶活性Dihydrofolate reductase activity/hymidylate synthase activity | dTMP生物合成/单碳代谢dTMP biosynthetic process/one-carbon metabolic process | - | ||
umc2041~ umc22870 | 139 | GRMZM2G094497 | ATP结合ATP binding | ATP水解耦合质子转运/质子转运/ATP代谢过程ATP hydrolysis coupled proton transport/proton transport/ATP metabolic process | 质子运输V型ATP酶/V1结构域Proton-transporting V-type ATPase/V1domain |
| GRMZM2G074122 | 催化活性/苯醇丙酮酸羧化酶活性Catalytic activity/hosphoenolpyruvate carboxylase activity | 三羧酸循环/碳固定Tricarboxylic acid cycle/carbon fixation | - | ||
| GRMZM2G162434 | DNA结合DNA binding | - | - | ||
| GRMZM2G116079 | 金属离子结合 Metalion binding | - | - | ||
umc2040~ bnlg1174a | 24 | GRMZM2G039113 | - | - | 细胞质/细胞骨架Cytoplsm/cytoskeleton |
umc2040~ bnlg1174a | 2 | GRMZM2G119169 | 核糖体结构组成Structural constituent of ribosome | 翻译Translation | 核糖体/核糖体大亚基/细胞内核糖体蛋白复合体Ribosome/large ribosomal subunit/intracellular ribonucleoprotein complex |
umc2040~ bnlg1174a | 48 | GRMZM2G163437 | ATP结合/葡萄糖-1-1磷酸腺苷转移酶活性ATP binding/glucose-1-phosphate adenylytransferase activity | 核糖体生物合成/淀粉生物合成Glycogen biosynthetic process/starch biosynthetic process | 叶绿体/质体Chloroplast/plastid |
sQTLs区间 sQTLs interval | 基因数量Number of gene | 基因 ID Gene ID | KEGG注释 KEGG annotation | Nr注释 Nr annotation (玉米Z. mays) | 功能 Function |
|---|---|---|---|---|---|
mmc0041~ phi308707 | 146 | GRMZM2G014392 | ABA/类胡萝卜素/次生代谢物生物合成Abscisic acid biosynthesis/carotenoids biosynthesis of secondary metabolites | Viviparous 14 | 水亏缺下ABA作用参与叶片生长Leaf growth via ABA under water deficit |
| GRMZM2G073725 | 氨基酸糖/核苷酸糖代谢Amino sugar and nucleotide sugar metabolism | 高尔基相关蛋白se-wap41 Golgi associated protein se-wap41 | 乙烯信号作用下参与干旱胁迫反应Drought stress response via ethylene signal | ||
| GRMZM2G017087 | 氨基酸代谢Metabolism of amino acids | Knotted 1 | 维持分生组织稳态/促进叶片形成Meristem homeostasis/leaf formation | ||
bnlg1233~ bnlg1520 | 104 | GRMZM2G140667 | 抗坏血酸和醛糖代谢/谷胱甘肽代谢Ascorbate and aldarate metabolism/glutathione metabolism | 抗坏血酸盐过氧化物酶2 Ascorbate peroxidase 2 | 干旱胁迫下维持叶片叶绿素量Chlorophyll content was maintained under drought stress |
| GRMZM2G005990 | 叶酸生物合成/四氢叶酸生物合成Folate biosynthesis/tetrahydrofolate biosynthesis | 双功能二氢叶酸还原酶-胸腺酸合成酶Bifunctional dihydrofolate reductase-thymidylate synthase | 碳代谢Carbon metabolism | ||
umc2041~ umc2287 | 139 | GRMZM2G094497 | 代谢途径/氧化磷酸化Metabolic pathways/oxidative phosphorylation | 空泡ATP合酶B亚基Vacuolar ATP synthase subunit B | 液泡ATP合酶亚基B Vacuolar ATP synthase subunit B |
| GRMZM2G074122 | C4 -二羧酸循环/NAD-苹果酸酶代谢C4-dicarboxylic acid cycle/NAD-malic enzyme type | 磷酸烯醇丙酮酸羧化酶1亚型Phosphoenolpyruvate carboxylase isoform 1 | 光合作用/C4植物中参与固碳作用Photosynthesis/carbon fixation in C4 plants | ||
| GRMZM2G162434 | 组织特异性生物系统Organism-specific biosystem | 转录因子MYB30 Transcription factor MYB30 | 生长发育/代谢调控/细胞形态/胁迫应答Growth and development/metabolic regulation/cell morphology/stress response | ||
| GRMZM2G116079 | 细胞色素代谢外源性物质Metabolism of xenobiotics by cytochrome | 假定的锌指蛋白Putative zinc finger protein30 | 调控细胞分化Cell differentiation | ||
umc2040~ bnlg1174a | 24 | GRMZM2G039113 | 囊泡运输SNARE作用SNARE interactions in vesicular transport | Tangled 1 | 细胞骨架排列/细胞分裂/叶片发育Cytoskeletal arrangement/cell division/leaf development |
bnlg1863~ umc2075 | 2 | GRMZM2G119169 | 核糖体生物合成、组织特异性生物系统Ribosome biosynthesis/organism-specific biosystem | 60S核糖体蛋白L17 60S ribosomal protein L17 | 组织特异性表达Tissue specific expression |
bnlg1655~ umc1345 | 48 | GRMZM2G163437 | 氨基酸/核苷酸糖代谢Amino sugar and nucleotide sugar metabolism | ADP葡糖糖焦磷酸化酶小亚基叶1 ADP glucose pyrophosphorylase small subunit leaf 1 | 促进光合作用和碳代谢Photosynthesis and carbon metabolism |
表5 叶面积sQTLs区间内候选基因检测及功能注释
Table 5 Candidate genes were detected and corresponding function were annotated in sQTLs intervals for leaf area
sQTLs区间 sQTLs interval | 基因数量Number of gene | 基因 ID Gene ID | KEGG注释 KEGG annotation | Nr注释 Nr annotation (玉米Z. mays) | 功能 Function |
|---|---|---|---|---|---|
mmc0041~ phi308707 | 146 | GRMZM2G014392 | ABA/类胡萝卜素/次生代谢物生物合成Abscisic acid biosynthesis/carotenoids biosynthesis of secondary metabolites | Viviparous 14 | 水亏缺下ABA作用参与叶片生长Leaf growth via ABA under water deficit |
| GRMZM2G073725 | 氨基酸糖/核苷酸糖代谢Amino sugar and nucleotide sugar metabolism | 高尔基相关蛋白se-wap41 Golgi associated protein se-wap41 | 乙烯信号作用下参与干旱胁迫反应Drought stress response via ethylene signal | ||
| GRMZM2G017087 | 氨基酸代谢Metabolism of amino acids | Knotted 1 | 维持分生组织稳态/促进叶片形成Meristem homeostasis/leaf formation | ||
bnlg1233~ bnlg1520 | 104 | GRMZM2G140667 | 抗坏血酸和醛糖代谢/谷胱甘肽代谢Ascorbate and aldarate metabolism/glutathione metabolism | 抗坏血酸盐过氧化物酶2 Ascorbate peroxidase 2 | 干旱胁迫下维持叶片叶绿素量Chlorophyll content was maintained under drought stress |
| GRMZM2G005990 | 叶酸生物合成/四氢叶酸生物合成Folate biosynthesis/tetrahydrofolate biosynthesis | 双功能二氢叶酸还原酶-胸腺酸合成酶Bifunctional dihydrofolate reductase-thymidylate synthase | 碳代谢Carbon metabolism | ||
umc2041~ umc2287 | 139 | GRMZM2G094497 | 代谢途径/氧化磷酸化Metabolic pathways/oxidative phosphorylation | 空泡ATP合酶B亚基Vacuolar ATP synthase subunit B | 液泡ATP合酶亚基B Vacuolar ATP synthase subunit B |
| GRMZM2G074122 | C4 -二羧酸循环/NAD-苹果酸酶代谢C4-dicarboxylic acid cycle/NAD-malic enzyme type | 磷酸烯醇丙酮酸羧化酶1亚型Phosphoenolpyruvate carboxylase isoform 1 | 光合作用/C4植物中参与固碳作用Photosynthesis/carbon fixation in C4 plants | ||
| GRMZM2G162434 | 组织特异性生物系统Organism-specific biosystem | 转录因子MYB30 Transcription factor MYB30 | 生长发育/代谢调控/细胞形态/胁迫应答Growth and development/metabolic regulation/cell morphology/stress response | ||
| GRMZM2G116079 | 细胞色素代谢外源性物质Metabolism of xenobiotics by cytochrome | 假定的锌指蛋白Putative zinc finger protein30 | 调控细胞分化Cell differentiation | ||
umc2040~ bnlg1174a | 24 | GRMZM2G039113 | 囊泡运输SNARE作用SNARE interactions in vesicular transport | Tangled 1 | 细胞骨架排列/细胞分裂/叶片发育Cytoskeletal arrangement/cell division/leaf development |
bnlg1863~ umc2075 | 2 | GRMZM2G119169 | 核糖体生物合成、组织特异性生物系统Ribosome biosynthesis/organism-specific biosystem | 60S核糖体蛋白L17 60S ribosomal protein L17 | 组织特异性表达Tissue specific expression |
bnlg1655~ umc1345 | 48 | GRMZM2G163437 | 氨基酸/核苷酸糖代谢Amino sugar and nucleotide sugar metabolism | ADP葡糖糖焦磷酸化酶小亚基叶1 ADP glucose pyrophosphorylase small subunit leaf 1 | 促进光合作用和碳代谢Photosynthesis and carbon metabolism |
环境 Environment | 时期 Stage | QTL (i) | 区间 Interval (i) | Bin (i) | QTL (j) | 区间 Interval (j) | Bin (j) | AA | h2 (AA) (%) |
|---|---|---|---|---|---|---|---|---|---|
| E6 | R1 | qLS-Ch.1-1 | mmc0041~phi308707 | 1.08~1.10 | qLS-J2-1 | bnlg1233~bnlg1520 | 2.08~2.09 | -0.68 | 4.83 |
表6 多环境下F2∶3(POPCT)群体相应叶叶面积上位性QTLs分析
Table 6 Epistatic interactions among QTLs for leaf area were analyzed in F2∶3 (POPCT) populations under multiple environments
环境 Environment | 时期 Stage | QTL (i) | 区间 Interval (i) | Bin (i) | QTL (j) | 区间 Interval (j) | Bin (j) | AA | h2 (AA) (%) |
|---|---|---|---|---|---|---|---|---|---|
| E6 | R1 | qLS-Ch.1-1 | mmc0041~phi308707 | 1.08~1.10 | qLS-J2-1 | bnlg1233~bnlg1520 | 2.08~2.09 | -0.68 | 4.83 |

图5 玉米75个叶发育候选基因的氨基酸序列系统进化树基因后面标有**表示为本研究检测到的相应基因。The ** on the back of the gene indicated that the gene is the detected gene in this study.
Fig.5 Phylogenetic tree of 75 candidate genes for leaf development via corresponding amino acid sequence in maize
| 1 | An Y Q, Zhang J, Xi Z Y, et al. QTL mapping of leaf area for different leaf position in maize (Zea mays L.). Molecular Plant Breeding, 2016, 14(8): 2113-2120. |
| 安允权, 张君, 席章营, 等. 玉米不同位叶叶面积的QTL定位. 分子植物育种, 2016, 14(8): 2113-2120. | |
| 2 | Zhang Z L, Jiang F, Liu P F, et al. QTL mapping of ear leaf area in sweet corn. Hubei Agricultural Sciences, 2014, 53(7): 1502-1505. |
| 张资丽, 蒋锋, 刘鹏飞, 等. 甜玉米穗位叶面积QTL定位. 湖北农业科学, 2014, 53(7): 1502-1505. | |
| 3 | Zhao X Q, Lu Y T, Bai M X, et al. Response of maize genotypes with different plant architecture to drought tolerance. Acta Prataculturae Sinica, 2020, 29(2): 149-162. |
| 赵小强, 陆晏天, 白明兴, 等. 不同株型玉米基因型对干旱胁迫的响应分析. 草业学报, 2020, 29(2): 149-162. | |
| 4 | Huang C X, Zhang H J. Effect of different irrigation amounts on leaf area dynamics and grain yield of spring maize (Zea mays) in oasis region. Journal of Water Resources & Water Engineering, 2016, 27(4): 229-232, 240. |
| 黄彩霞, 张恒嘉. 不同灌水量对绿洲玉米叶面积动态及产量的影响. 水资源与水工程学报, 2016, 27(4): 229-232, 240. | |
| 5 | Zhao X Q, Fang P, Zhang J W, et al. QTL mapping for six ear leaf architecture traits under water-stressed and well-watered conditions in maize (Zea mays L.). Plant Breeding, 2018, 137(1): 60-72. |
| 6 | Maes Y H, Steppe K. Estimating evapotranspiration and drought stress with ground-based thermal remote sensing in agricultura: A review. Journal of Experimental Botany, 2015, 63: 4671-4712. |
| 7 | Ribaut J, Betran J, Mnneveux P, et al. Drought tolerance in maize//Handbook of maize: Its biology. New York: Springer, 2009: 311-344. |
| 8 | Pagano E, Cela S, Maddonni G A, et al. Intra-specific competition in maize: Ear development, flowering dynamics and kernel set of carly-established plant hierarchies. Field Crop Research, 2007, 102(3): 198-209. |
| 9 | Mickelson S M, Stuber C S, Senior L, et al. Quantitative trait loci controlling leaf and tassel trait in a B73×Mo17 population of maize. Crop Science, 2002, 42(6): 1902-1909. |
| 10 | Quarrie S A, Stojanovic J, Sofija P J. Improving drought resistance in small-grained cereals: A case study, progress and prospects. Plant growth Regulation, 1999, 29(1): 1-21. |
| 11 | Dong Y B, Zhang Z W, Shi Q L, et al. QTL consistency for agronomic traits across three generations and potential applications in popcorn. Journal of Integrative Agriculture, 2015, 14(12): 2547-2557. |
| 12 | Cai H G, Chu Q, Yuan L X, et al. Identification of quantitative trait loci for leaf area and chlorophyII content in maize (Zea mays) under low nitrogen and low phosphorus supply. Molecular Breeding, 2012, 30: 251-266. |
| 13 | Nikolic A, Andjelkovic V, Dodig D, et al. Quantitative trait loci for yield and morphological traits in maize under drought stress. Genetika, 2011, 43: 263-276. |
| 14 | Liu J C, Chu Q, Cai H G, et al. SSR linkage map construction and QTL mapping for leaf area in maize. Hereditas, 2010, 32(6): 625-631. |
| 刘建超, 褚群, 蔡红光, 等. 玉米SSR连锁图谱构建及叶面积的QTL定位. 遗传, 2010, 32(6): 625-631. | |
| 15 | Agrama H A S, Zakaria A G, Said F B, et al. Identification of quantitative trait loci for nitrogen use efficiency in maize. Molecular Breeding, 1999, 5(2): 187-195. |
| 16 | Zhao X Q, Ren B, Peng Y L, et al. Epistatic and QTL×environment interaction effects for ear related traits in two maize (Zea mays) populations under eight watering environments. Acta Agronomica Sinica, 2019, 45(6): 856-871. |
| 赵小强, 任斌, 彭云玲, 等. 8种水旱环境下2个玉米群体穗部性状QTL间的上位性及环境互作效应分析. 作物学报, 2019, 45(6): 856-871. | |
| 17 | Zhao X Q, Fang P, Peng Y L, et al. Genetic analysis and QTL mapping for seven agronomic traits in maize (Zea mays) using two connected populations. Acta Prataculturae Sinica, 2018, 27(9): 152-165. |
| 赵小强, 方鹏, 彭云玲, 等. 基于两个相关群体的玉米7个主要农艺性状遗传分析和QTL定位. 草业学报, 2018, 27(9): 152-165. | |
| 18 | Zhao X Q, Peng Y L, Zhang J W, et al. Mapping QTLs and meta-QTLs for two inflorescence architecture traits in multiple maize populations under different watering environments. Molecular Breeding, 2017, 37(7): 91. |
| 19 | Zhao X Q, Peng Y L, Li J Y, et al. Comprehensive evaluation of salt tolerance in 16 maize inbred lines. Agricultural Research in the Arid Areas, 2014, 32(5): 40-45, 51. |
| 赵小强, 彭云玲, 李建英, 等. 16份玉米自交系的耐盐性评价. 干旱地区农业研究, 2014, 32(5): 40-45, 51. | |
| 20 | Zhao X Q, Fang P, Peng Y L, et al. QTL mapping for six ear-related traits based on two maize (Zea mays) related populations. Journal of Agricultural Biotechnology, 2018, 26(5): 729-742. |
| 赵小强, 方鹏, 彭云玲, 等. 基于两个相关群体的玉米6个穗部性状QTL定位. 农业生物技术学报, 2018, 26(5): 729-742. | |
| 21 | Zhao X Q, Peng Y L, Zhang J W, et al. Identification of QTLs and meta-QTLs for seven agronomic traits in multiple maize populations under well-watered and water-stressed conditions. Crop Science, 2018, 58(2): 507-520. |
| 22 | Knapp S J, Stroup W W, Ross W M. Exact confidence intervals for heritability on a progeny mean basis. Crop Science, 1985, 25(1): 192-194. |
| 23 | Van-Ooijen J W, Join M. Software for the calculation of genetic linkage maps in experimental populations. The Netherlands: Kyazma Wageningen. (http://www.kyazma.nl/index.php/mc.JoinMap/sc.Evaluate/). |
| 24 | Churchill G A, Doerge R W. Empirical threshold values for quantitative trait mapping. Genetics, 1994, 138(3): 963-971. |
| 25 | Stuber C W, Edwards M D, Wendel J. F1 molecular marker facilitated investigations of quantitative trait loci in maize II. Factors influencing yield and its component traits. Crop Science, 1987, 27: 639-648. |
| 26 | McCouch S R, Cho Y G, Yano P E, et al. Report on QTL nomenclature. Rice Genet New Slett, 1997, 14: 11-13. |
| 27 | Yang J, Zhu J, Williams R W. Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics, 2007, 23(12): 1527-1536. |
| 28 | Tuberosa R, Salvi S, Sanguineti M C, et al. Mapping QTL regulating morpho-physiological traits and yields: Case studies, short comings and perspectives in drought-stress maize. Annals of Botany, 2002, 89(7): 941-963. |
| 29 | Ashbumer M. Gene ontology: Tool for the unifieation of biology. Nature Genet, 2000, 25(1): 25-29. |
| 30 | Kanehisa M, Goto S, Sato Y, et al. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res, 2014, 42: 199-205. |
| 31 | Guo S L, Zhang J, Qi J S, et al. Analysis of meta-quantitative trait loci and their candidate genes related to leaf shape in maize. Chinese Bulletin of Botany, 2018, 53(4): 487-501. |
| 郭书磊, 张君, 齐建双, 等. 玉米叶形相关性状的Meta-QTL及候选基因分析. 植物学报, 2018, 53(4): 487-501. | |
| 32 | Zhou D S, Zhao Y M, Yan M, et al. Analysis of genetic effect and genotype by environment of leaf area of different leaf position in maize. Chinese Agricultural Science Bulletin, 2008, 24(4): 195-198. |
| 周东升, 赵延明, 严敏, 等. 玉米不同叶位叶面积遗传效应及与环境互作效应分析. 中国农学通报, 2008, 24(4): 195-198. | |
| 33 | Ma J, Wang T G, Zhang H S, et al. Genetic analysis on three leaves area near the ear by mixed inheritance model of major genes and polygenes in maize. Journal of Henan Agricultural Sciences, 2012, 41(4): 25-28. |
| 马娟, 王铁固, 张怀胜, 等. 玉米穗三叶叶面积主基因+多基因遗传模型分析. 河南农业科学, 2012, 41(4): 25-28. | |
| 34 | Peng J, Cai Y L, Xu D L, et al. Genetic analysis of plant-type characters in maize (Zea mays L.) by using joint analysis of multiple generations. Journal of Biomathematics, 2009, 24(1): 149-156. |
| 彭静, 蔡一林, 徐德林, 等. 玉米株型性状多世代联合遗传分析. 生物数学学报, 2009, 24(1): 149-156. | |
| 35 | Si S L, Hao X J, Wei C, et al. The correlation and heterosis of plant-type traits in maize. Journal of Maize Sciences, 2009, 17(1): 51-53. |
| 司书丽, 郝学景, 魏春, 等. 玉米株型性状的亲子相关与杂种优势. 玉米科学, 2009, 17(1): 51-53. | |
| 36 | Xu C, Wang B, Mao K J, et al. QTL mapping for plant-type related traits using single segment substitution lines in maize. Journal of Maize Sciences, 2014, 22(2): 28-34. |
| 许诚, 王彬, 毛克举, 等. 利用单片段代换系群体定位玉米株型性状QTL. 玉米科学, 2014, 22(2): 28-34. | |
| 37 | Tang J H, Teng W T, Yan J. Genetic dissection of plant height by molecular by molecular markers using a population of recombinant inbred lines in maize. Euphytica, 2007, 155: 117-124. |
| 38 | Zheng Z P, Huang Y B, Tian M L, et al. Mapping QTLs and epistasis for plant type traits in maize under two nitrogen levels. Journal of Maize Sciences, 2007, 15(2): 14-18. |
| 郑祖平, 黄玉碧, 田孟良, 等. 不同供氮水平下玉米株型相关性状的QTLs定位和上位性效应分析. 玉米科学, 2007, 15(2): 14-18. | |
| 39 | Pelleschi S, Leonardi A, Rocher J P, et al. Analysis of the relationships between growth, photosynthesis and carbohydrate metabolism using quantitative trait loci (QTLs) in young maize plants subjected to water deprivation. Molecular Breeding, 2006, 17: 21-39. |
| 40 | Voisin A S, Reidy B, Parent B, et al. Are ABA, ethylene or their interaction involved in the response of leaf growth to soil water deficit? An analysis using naturally occurring variation or genetic transformation of ABA production in maize. Plant Cell Environ, 2006, 29(9): 1829-1840. |
| 41 | Jia J P, Fu J J, Zheng J, et al. Annotation and expression profile analysis of 2073 full-length cDNAs from stress-induced maize (Zea mays L.) seedlings. Plant Journal, 2006, 48(5): 710-727. |
| 42 | Ramirez J, Bolduc N, Lisch D, et al. Distal expression of knotted in maize leaves leads to reestablishment of proximal/distal patterning and leaf dissection. Physiologia Plantarum, 2009, 151(4): 1878-1888. |
| 43 | Lunde C, Hake S. The interaction of knotted1 and thick tassel dwarf1 in vegetative and reproductive meristems of maize. 2009, 181(4): 1693-1697. |
| 44 | Singh D G, Lomako J, Lomako W M, et al. Beta-glucosylarginine: A new glucose-protein bond in a self-glucosylating protein from sweet corn. Febs Letters, 1995, 376(1): 61-64. |
| 45 | Zhao N, Zhao F, Li Y H. Advances in research on zinc finger protein. Letters in Biotechnology, 2009, 20(1): 131-134. |
| 赵楠, 赵飞, 李玉花. 锌指蛋白结构及功能研究进展. 生物技术通讯, 2009, 20(1): 131-134. | |
| 46 | Cleary A L, Smith L G. The tangled1 gene is required for spatial control of cytoskeletal arrays associated with cell division during maize leaf development. Plant Cell, 1998, 10(11): 1875-1888. |
| 47 | Smidansky E D, Meyer F D, Blakeslee B, et al. Expression of a modified ADP-glucose pyrophosphorylase large subunit in wheat seeds stimulates photosynthesis and carbon metabolism. Planta, 2007, 225(4): 965-976. |
| 48 | Xing W H, Li T, Qiao Q, et al. The QTL mapping for leaf area based on maize Ye478 introgression lines. Molecular Plant Breeding, 2019, 17(6): 1938-1943. |
| 邢文慧, 李彤, 乔巧, 等. 基于掖478导入系的玉米叶面积QTL定位. 分子植物育种, 2019, 17(6): 1938-1943. | |
| 49 | Zhao X Q. Genetic mechanisms study of drought tolerance related to plant architecture in maize (Zea mays L.). Lanzhou: Gansu Agricultural University, 2018. |
| 赵小强. 玉米株型相关耐旱遗传机理研究. 兰州: 甘肃农业大学, 2018. | |
| 50 | Li X T, Ding J Q, Wang R X, et al. QTL mapping of related traits of plant type in maize. Jiangsu Agricultural Sciences, 2011, 39(2): 21-25. |
| 李贤唐, 丁俊强, 王瑞霞, 等. 玉米株型相关性状的QTL定位与分析. 江苏农业科学, 2011, 39(2): 21-25. |
| [1] | 黄丽琴, 李松桥, 袁振中, 唐晶, 闫景彩, 唐启源. 全株水稻与平菇菌糠共发酵料对浏阳黑山羊屠宰性能、肉品质和器官指数的影响[J]. 草业学报, 2021, 30(6): 133-140. |
| [2] | 祁鹤兴, 芦光新, 李宗仁, 徐成体, 德科加, 周孝娟, 王英成, 马桂花. 青海省青贮玉米链格孢叶枯病病原菌鉴定及其致病力分析[J]. 草业学报, 2021, 30(6): 94-105. |
| [3] | 臧真凤, 白婕, 刘丛, 昝看卓, 龙明秀, 何树斌. 紫花苜蓿形态和生理指标响应干旱胁迫的品种特异性[J]. 草业学报, 2021, 30(6): 73-81. |
| [4] | 罗巧玉, 王彦龙, 陈志, 马永贵, 任启梅, 马玉寿. 水分逆境对发草脯氨酸及其代谢途径的影响[J]. 草业学报, 2021, 30(5): 75-83. |
| [5] | 候怡谣, 李霄, 龙瑞才, 杨青川, 康俊梅, 郭长虹. 过量表达紫花苜蓿MsHB7基因对拟南芥耐旱性的影响[J]. 草业学报, 2021, 30(4): 170-179. |
| [6] | 王子欣, 胡国铮, 水宏伟, 葛怡情, 韩玲, 高清竹, 干珠扎布, 旦久罗布. 不同时期干旱对青藏高原高寒草甸生态系统碳交换的影响[J]. 草业学报, 2021, 30(4): 24-33. |
| [7] | 张宁, 曹允馨, 徐伟, 常智慧. 干旱胁迫下污泥对草地早熟禾生长及激素代谢的影响[J]. 草业学报, 2021, 30(3): 167-176. |
| [8] | 刘凯强, 刘文辉, 贾志锋, 梁国玲, 马祥. 干旱胁迫对‘青燕1号’燕麦产量及干物质积累与分配的影响[J]. 草业学报, 2021, 30(3): 177-188. |
| [9] | 罗文蓉, 胡国铮, 干珠扎布, 高清竹, 李岩, 葛怡情, 李钰, 何世丞, 旦久罗布. 模拟干旱对藏北高寒草甸植物物候期和生产力的影响[J]. 草业学报, 2021, 30(2): 82-92. |
| [10] | 李冬, 申洪涛, 王艳芳, 王悦华, 王丽君, 赵世民, 刘领. 外源褪黑素对干旱胁迫下烟草幼苗光合碳同化和内源激素的影响[J]. 草业学报, 2021, 30(1): 130-139. |
| [11] | 刘桃桃, 王思伟, 李秋凤, 曹玉凤, 王昆, 王丽娟, 沈宜钊, 孙雪丽, 张美琦, 闫金玲, 李建国, 高艳霞, 王美美. 利用尼龙袋法比较3个全株玉米品种青贮前后肉牛瘤胃降解特性[J]. 草业学报, 2021, 30(1): 159-169. |
| [12] | 李振松, 万里强, 李硕, 李向林. 苜蓿根系构型及生理特性对干旱复水的响应[J]. 草业学报, 2021, 30(1): 189-196. |
| [13] | 郭家萌, 何灵芝, 闫东良, 李卓, 王泳超, 邵瑞鑫, 杨青华. 控释氮肥和尿素配比对不同品种夏玉米氮素累积、转移及其利用效率的影响[J]. 草业学报, 2021, 30(1): 81-95. |
| [14] | 齐鹏, 王晓娇, 姚一铭, 陈晓龙, 武均, 蔡立群. 不同耕作方法和施氮量对旱作农田土壤CO2排放及碳平衡的影响[J]. 草业学报, 2021, 30(1): 96-106. |
| [15] | 雷恩, 邵迪, 朱天彪, 舒星, 杨永兵, 王岳东, 唐启源. 饲用玉米器官含水率、力学强度与籽粒机收质量的关系研究[J]. 草业学报, 2020, 29(9): 125-135. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||